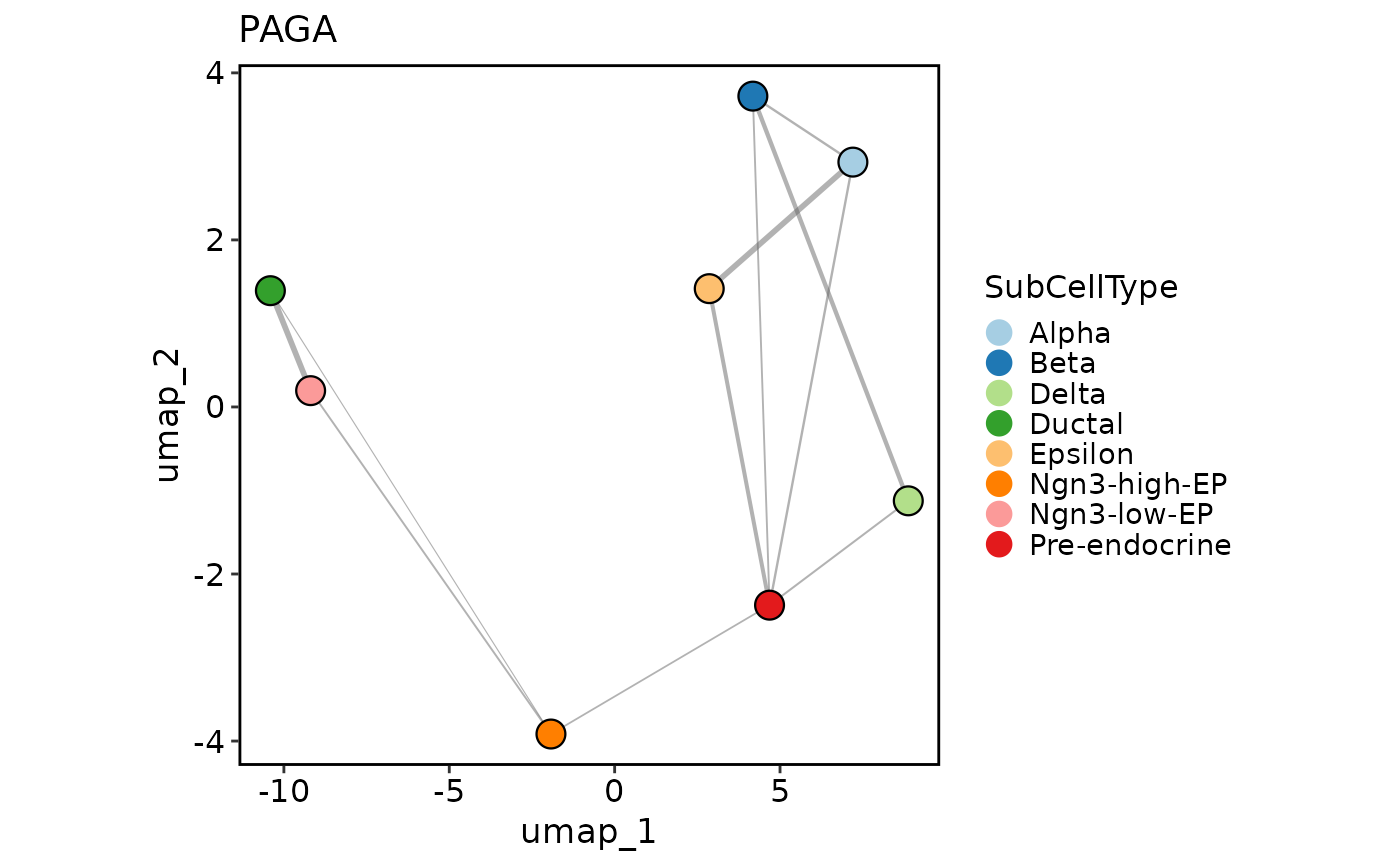
PAGA is a graph-based method used to infer cellular trajectories. This function runs the PAGA analysis on a Seurat object.
Usage
RunPAGA(
srt = NULL,
adata = NULL,
assay_x = "RNA",
layer_x = "counts",
assay_y = c("spliced", "unspliced"),
layer_y = "counts",
group.by = NULL,
linear_reduction = NULL,
nonlinear_reduction = NULL,
basis = NULL,
n_pcs = 30,
n_neighbors = 30,
use_rna_velocity = FALSE,
vkey = "stochastic",
embedded_with_PAGA = FALSE,
paga_layout = "fr",
threshold = 0.1,
point_size = 20,
infer_pseudotime = FALSE,
root_group = NULL,
root_cell = NULL,
n_dcs = 10,
n_branchings = 0,
min_group_size = 0.01,
palette = "Chinese",
palcolor = NULL,
legend.position = "on data",
cores = 1,
show_plot = FALSE,
save_plot = FALSE,
plot_format = c("pdf", "png", "svg"),
plot_dpi = 300,
plot_prefix = "paga",
dirpath = "./paga",
backend = c("python", "cpp"),
return_seurat = !is.null(srt),
verbose = TRUE
)Arguments
- srt
A Seurat object. Default is
NULL. If provided,adatawill be ignored.- adata
An anndata object. Default is
NULL.- assay_x
Assay to convert as the main data matrix in the anndata object. Default is
"RNA".- layer_x
Layer name for assay_x in the Seurat object. Default is
"counts".- assay_y
Assays to convert as layers in the anndata object. Default is
c("spliced", "unspliced").- layer_y
Layer names for the assay_y in the Seurat object. Default is
"counts".- group.by
Name of one or more meta.data columns to group (color) cells by.
- linear_reduction
The linear dimensionality reduction method to use. Options are
"pca","svd","ica","nmf","mds", or"glmpca". Default is"pca".- nonlinear_reduction
The nonlinear dimensionality reduction method to use. Options are
"umap","umap-naive","tsne","dm","phate","pacmap","trimap","largevis", or"fr". Default is"umap".- basis
The basis to use for reduction, e.g.,
"UMAP".- n_pcs
Number of principal components to use for linear reduction. Default is
30.- n_neighbors
Number of neighbors to use for constructing the KNN graph. Default is
30.- use_rna_velocity
Whether to use RNA velocity for PAGA analysis. Default is
FALSE.- vkey
The name of the RNA velocity data to use if
use_rna_velocityisTRUE. Default is"stochastic".- embedded_with_PAGA
Whether to embed data using PAGA layout. Default is
FALSE.- paga_layout
The layout for plotting PAGA graph. See layout param in
scanpy.pl.pagafunction.- threshold
The threshold for plotting PAGA graph. Edges for weights below this threshold will not be drawn.
- point_size
The point size for plotting.
- infer_pseudotime
Whether to infer pseudotime.
- root_group
The group to use as the root for pseudotime inference.
- root_cell
The cell to use as the root for pseudotime inference.
- n_dcs
The number of diffusion components to use for pseudotime inference.
- n_branchings
Number of branchings to detect.
- min_group_size
The minimum size of a group (as a fraction of the total number of cells) to consider it as a potential branching point.
- palette
Color palette name. Available palettes can be found in thisplot::show_palettes. Default is
"Chinese".- palcolor
Custom colors used to create a color palette. Default is
NULL.- legend.position
Position of legend in plots. Can be
"on data","right margin","bottom right", etc. Default is"on data".- cores
The number of cores to use for
cellrank.- show_plot
Whether to show the plot. Default is
FALSE.- save_plot
Whether to save plots to files. Default is
FALSE.- plot_format
Format for saved plots:
"png"(default),"pdf", or"svg".- plot_dpi
Resolution (DPI) for saved plots. Default is
300.- plot_prefix
Prefix for saved plot filenames. Default is "cellrank".
- dirpath
The directory to save the plots. Default is
"./cellrank".- backend
Backend used to compute PAGA.
"python"keeps the original scanpy workflow and remains the default."cpp"uses the native C++ implementation for the standard connectivity graph and tree, plus an approximate R igraph layout stored inpaga$pos.- return_seurat
Whether to return a Seurat object instead of an anndata object. Default is
TRUE.- verbose
Whether to print the message. Default is
TRUE.
Examples
data(pancreas_sub)
pancreas_sub <- standard_scop(pancreas_sub)
#> ℹ [2026-07-02 09:46:59] Start standard processing workflow...
#> ℹ [2026-07-02 09:46:59] Checking a list of <Seurat>...
#> ! [2026-07-02 09:47:00] Data 1/1 of the `srt_list` is "unknown"
#> ℹ [2026-07-02 09:47:00] Perform `NormalizeData()` with `normalization.method = 'LogNormalize'` on 1/1 of `srt_list`...
#> ℹ [2026-07-02 09:47:00] Perform `FindVariableFeatures()` on 1/1 of `srt_list`...
#> ℹ [2026-07-02 09:47:00] Use the separate HVF from `srt_list`
#> ℹ [2026-07-02 09:47:00] Number of available HVF: 2000
#> ℹ [2026-07-02 09:47:00] Finished check
#> ℹ [2026-07-02 09:47:00] Perform `ScaleData()`
#> ℹ [2026-07-02 09:47:00] Perform pca linear dimension reduction
#> ℹ [2026-07-02 09:47:01] Use stored estimated dimensions 1:23 for Standardpca
#> ℹ [2026-07-02 09:47:01] Perform `Seurat::FindClusters()` with `cluster_algorithm = 'louvain'` and `cluster_resolution = 0.6`
#> ℹ [2026-07-02 09:47:01] Reorder clusters...
#> ℹ [2026-07-02 09:47:03] Skip `log1p()` because `layer = data` is not "counts"
#> ℹ [2026-07-02 09:47:03] Perform umap nonlinear dimension reduction
#> ✔ [2026-07-02 09:47:10] Standard processing workflow completed
pancreas_sub <- RunPAGA(
pancreas_sub,
assay_x = "RNA",
group.by = "SubCellType",
linear_reduction = "PCA",
nonlinear_reduction = "UMAP",
backend = "cpp"
)
#> ℹ [2026-07-02 09:47:10] Running PAGA with BiocNeighbors using 29 neighbors
#> ✔ [2026-07-02 09:47:10] PAGA cpp backend completed
PAGAPlot(pancreas_sub, reduction = "UMAP")
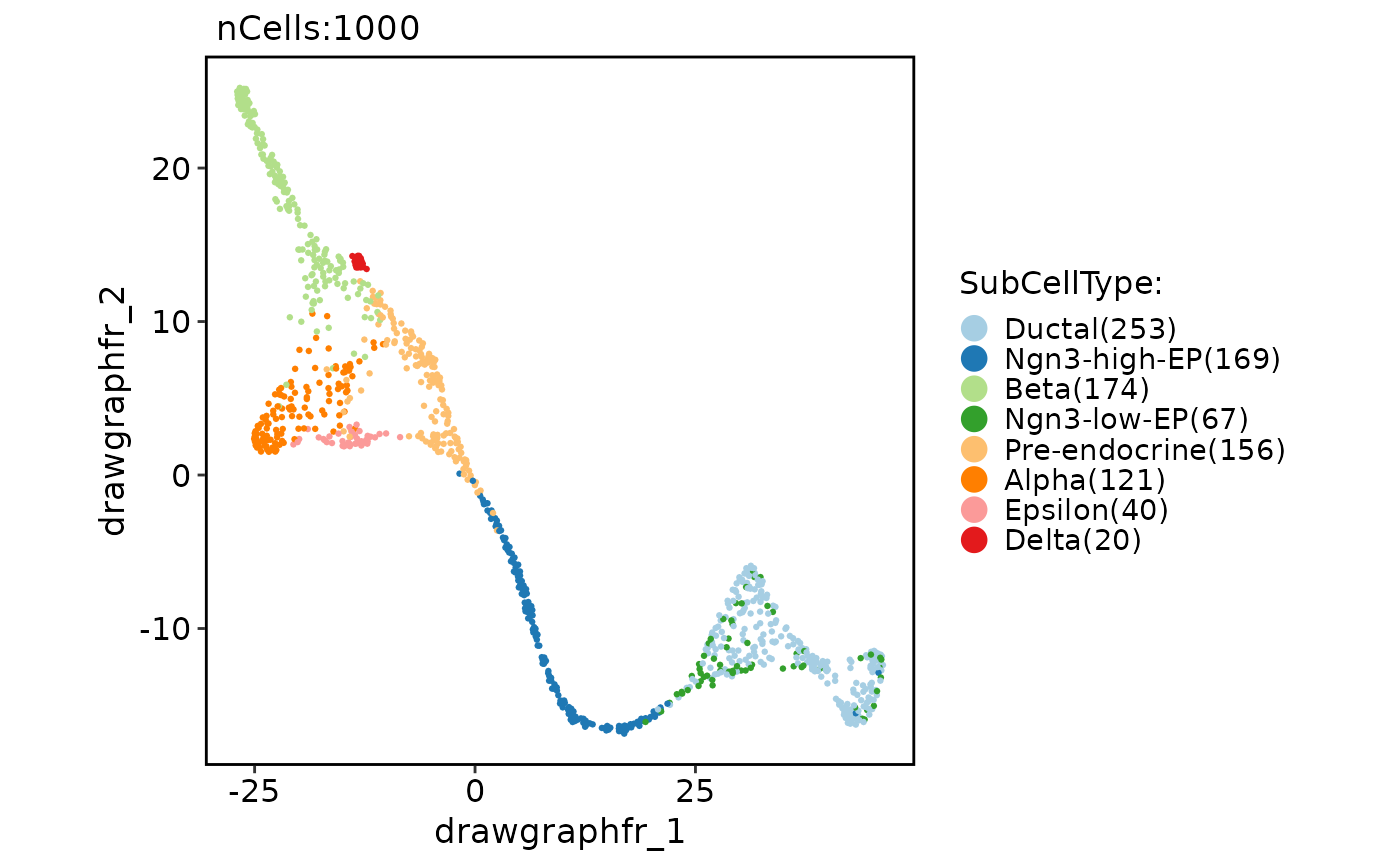 CellDimPlot(
pancreas_sub,
group.by = "SubCellType",
reduction = "UMAP",
paga = pancreas_sub@misc$paga
)
CellDimPlot(
pancreas_sub,
group.by = "SubCellType",
reduction = "UMAP",
paga = pancreas_sub@misc$paga
)