This function performs cell scoring on a Seurat object.
It calculates scores for a given set of features and stores them in meta.data
and/or a score assay, depending on new_assay and store_metadata.
Usage
CellScoring(
srt,
features = NULL,
layer = "data",
assay = NULL,
split.by = NULL,
IDtype = "symbol",
species = "Homo_sapiens",
db = "GO_BP",
termnames = NULL,
db_update = FALSE,
db_version = "latest",
convert_species = TRUE,
Ensembl_version = NULL,
mirror = NULL,
minGSSize = 10,
maxGSSize = 500,
method = "Seurat",
backend = c("cpp", "r"),
cpp_strategy = c("sparse", "aucell", "topk", "full"),
classification = TRUE,
name = "",
new_assay = FALSE,
store_metadata = NULL,
seed = 11,
cores = 1,
verbose = TRUE,
...
)Arguments
- srt
A Seurat object.
- features
A named list of feature lists for scoring. If
NULL,dbwill be used to create features sets.- layer
Which layer to use. Default is
data.- assay
Which assay to use. If
NULL, the default assay of the Seurat object will be used. When the object also containsChromatinAssay, the default assay and additionalChromatinAssaywill be preprocessed sequentially.- split.by
Name of a column in meta.data column to split plot by. Default is
NULL.- IDtype
A character vector specifying the type of gene IDs in the
srtobject orgeneIDargument. This argument is used to convert the gene IDs to a different type ifIDtypeis different fromresult_IDtype.- species
A character vector specifying the species for which the gene annotation databases should be prepared. Can be
"Homo_sapiens"or"Mus_musculus".- db
A character vector specifying the annotation sources to be included in the gene annotation databases. Can be one or more of
"GO", "GO_BP", "GO_CC", "GO_MF", "KEGG", "WikiPathway", "Reactome", "CORUM", "MP", "DO", "HPO", "PFAM", "CSPA", "Surfaceome", "SPRomeDB", "VerSeDa", "TFLink", "hTFtarget", "TRRUST", "JASPAR", "ENCODE", "MSigDB", "CellTalk", "CellChat", "Chromosome", "GeneType", "Enzyme", "TF", "CytoTRACE2". MSigDB subcollections can be requested as"MSigDB_<collection>", such as"MSigDB_H"for human Hallmark and"MSigDB_MH"for mouse Hallmark. Note:"CytoTRACE2"is species-independent and downloads pre-trained model data required by RunCytoTRACE.- termnames
A vector of term names to be used from the database. Default is
NULL, in which case all features from the database are used.- db_update
Whether the gene annotation databases should be forcefully updated. If set to FALSE, the function will attempt to load the cached databases instead. Default is
FALSE.- db_version
A character vector specifying the version of the gene annotation databases to be retrieved. Default is
"latest".- convert_species
Whether to use a species-converted database when the annotation is missing for the specified species. Default is
TRUE.- Ensembl_version
An integer specifying the Ensembl version. Default is
NULL. IfNULL, the latest version will be used.- mirror
Specify an Ensembl mirror to connect to. The valid options here are
"www","uswest","useast","asia".- minGSSize
The minimum size of a gene set to be considered in the enrichment analysis.
- maxGSSize
The maximum size of a gene set to be considered in the enrichment analysis.
- method
The method to use for scoring. Can be
"Seurat","AUCell","UCell","GSVA","ssGSEA","zscore","PLAGE", or"VISION". Multiple methods can be supplied at once; in that case each method is run separately and stored with a method suffix such as"GO_AUCell"or"GO_GSVA". Default is"Seurat".- backend
Scoring backend.
"cpp"is the default for supported methods."r"uses the original package implementation."cpp"currently supportsmethod = "Seurat",method = "AUCell",method = "GSVA",method = "ssGSEA",method = "zscore", andmethod = "PLAGE".method = "UCell"andmethod = "VISION"fall back to"r"whenbackendis not explicitly set.- cpp_strategy
AUCell scoring strategy used when
backend = "cpp"."sparse"ranks non-zero genes and approximates zero ties without densifying the expression matrix."aucell"calls the officialAUCell::AUCell_buildRankings()andAUCell::AUCell_calcAUC()path for exact consistency with the R backend,"topk"ranks only genes that can contribute to AUCell AUC, and"full"ranks all genes.- classification
Whether to perform classification based on the scores. Default is
TRUE.- name
The name of the assay to store the scores in. Only used if new_assay is TRUE. Default is
"".- new_assay
Whether to create a new assay for storing the scores. Default is
FALSE.- store_metadata
Whether to also store score columns in
meta.data. WhenNULL, manualfeatures = list(...)input is stored inmeta.databy default, while database-derived results stay assay-only whennew_assay = TRUE.- seed
Random seed for reproducibility. Default is
11.- cores
The number of cores to use for parallelization with foreach::foreach. Default is
1.- verbose
Whether to print the message. Default is
TRUE.- ...
Additional arguments to be passed to the scoring methods.
Examples
data(pancreas_sub)
pancreas_sub <- standard_scop(pancreas_sub)
#> ℹ [2026-07-02 08:28:44] Start standard processing workflow...
#> ℹ [2026-07-02 08:28:44] Checking a list of <Seurat>...
#> ! [2026-07-02 08:28:44] Data 1/1 of the `srt_list` is "unknown"
#> ℹ [2026-07-02 08:28:44] Perform `NormalizeData()` with `normalization.method = 'LogNormalize'` on 1/1 of `srt_list`...
#> ℹ [2026-07-02 08:28:45] Perform `FindVariableFeatures()` on 1/1 of `srt_list`...
#> ℹ [2026-07-02 08:28:45] Use the separate HVF from `srt_list`
#> ℹ [2026-07-02 08:28:45] Number of available HVF: 2000
#> ℹ [2026-07-02 08:28:45] Finished check
#> ℹ [2026-07-02 08:28:45] Perform `ScaleData()`
#> ℹ [2026-07-02 08:28:45] Perform pca linear dimension reduction
#> ℹ [2026-07-02 08:28:46] Use stored estimated dimensions 1:23 for Standardpca
#> ℹ [2026-07-02 08:28:46] Perform `Seurat::FindClusters()` with `cluster_algorithm = 'louvain'` and `cluster_resolution = 0.6`
#> ℹ [2026-07-02 08:28:46] Reorder clusters...
#> ℹ [2026-07-02 08:28:46] Skip `log1p()` because `layer = data` is not "counts"
#> ℹ [2026-07-02 08:28:46] Perform umap nonlinear dimension reduction
#> ✔ [2026-07-02 08:28:50] Standard processing workflow completed
features_all <- rownames(pancreas_sub)
pancreas_sub <- CellScoring(
pancreas_sub,
features = list(
A = features_all[1:100],
B = features_all[101:200]
),
method = "AUCell",
name = "test"
)
#> ℹ [2026-07-02 08:28:50] Start cell scoring
#> ℹ [2026-07-02 08:28:50] Data type is log-normalized
#> ℹ [2026-07-02 08:28:50] Number of feature lists to be scored: 2
#> ✔ [2026-07-02 08:28:51] Cell scoring completed
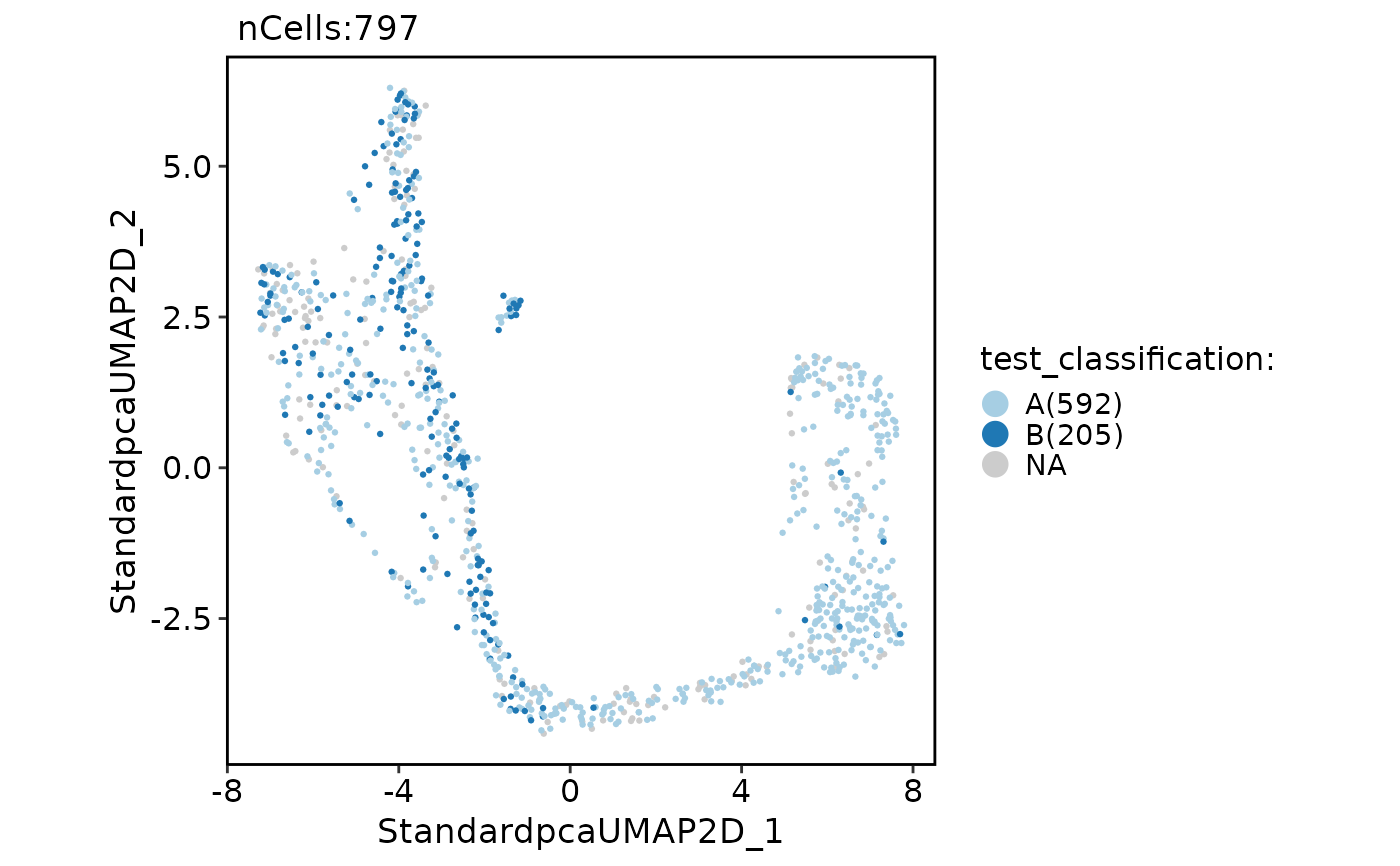
CellDimPlot(pancreas_sub, "test_classification")
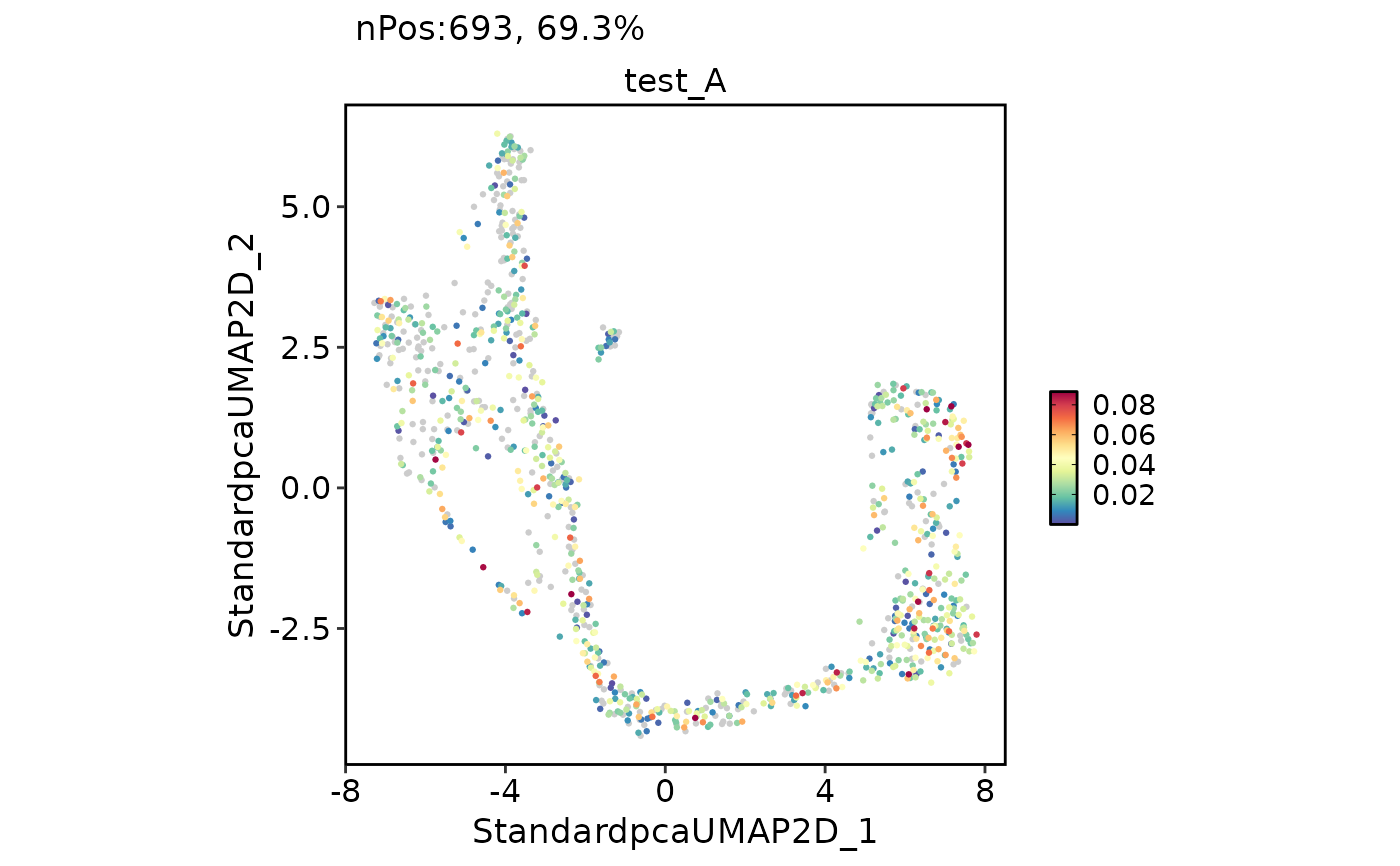
 FeatureDimPlot(
pancreas_sub,
features = "test_A"
)
FeatureDimPlot(
pancreas_sub,
features = "test_A"
)
 pancreas_sub <- CellScoring(
pancreas_sub,
features = list(A = features_all[1:100]),
method = c("AUCell", "GSVA")
)
#> ℹ [2026-07-02 08:28:51] Start cell scoring
#> ℹ [2026-07-02 08:28:51] Start cell scoring
#> ℹ [2026-07-02 08:28:51] Data type is log-normalized
#> ℹ [2026-07-02 08:28:52] Number of feature lists to be scored: 1
#> ✔ [2026-07-02 08:28:52] Cell scoring completed
#> ℹ [2026-07-02 08:28:52] Start cell scoring
#> ℹ [2026-07-02 08:28:52] Data type is log-normalized
#> ℹ [2026-07-02 08:28:52] Number of feature lists to be scored: 1
#> ℹ 2516278 nonzeros (less than 2^31) and 84.27% sparsity
#> ✔ [2026-07-02 08:30:17] Cell scoring completed
FeatureStatPlot(
pancreas_sub,
stat.by = c("AUCell_A", "GSVA_A"),
group.by = "CellType",
plot.by = "feature",
plot_type = "violin",
stack = TRUE
)
#> ℹ [2026-07-02 08:30:17] Setting `group.by` to "Features" as `plot.by` is set to "feature"
pancreas_sub <- CellScoring(
pancreas_sub,
features = list(A = features_all[1:100]),
method = c("AUCell", "GSVA")
)
#> ℹ [2026-07-02 08:28:51] Start cell scoring
#> ℹ [2026-07-02 08:28:51] Start cell scoring
#> ℹ [2026-07-02 08:28:51] Data type is log-normalized
#> ℹ [2026-07-02 08:28:52] Number of feature lists to be scored: 1
#> ✔ [2026-07-02 08:28:52] Cell scoring completed
#> ℹ [2026-07-02 08:28:52] Start cell scoring
#> ℹ [2026-07-02 08:28:52] Data type is log-normalized
#> ℹ [2026-07-02 08:28:52] Number of feature lists to be scored: 1
#> ℹ 2516278 nonzeros (less than 2^31) and 84.27% sparsity
#> ✔ [2026-07-02 08:30:17] Cell scoring completed
FeatureStatPlot(
pancreas_sub,
stat.by = c("AUCell_A", "GSVA_A"),
group.by = "CellType",
plot.by = "feature",
plot_type = "violin",
stack = TRUE
)
#> ℹ [2026-07-02 08:30:17] Setting `group.by` to "Features" as `plot.by` is set to "feature"
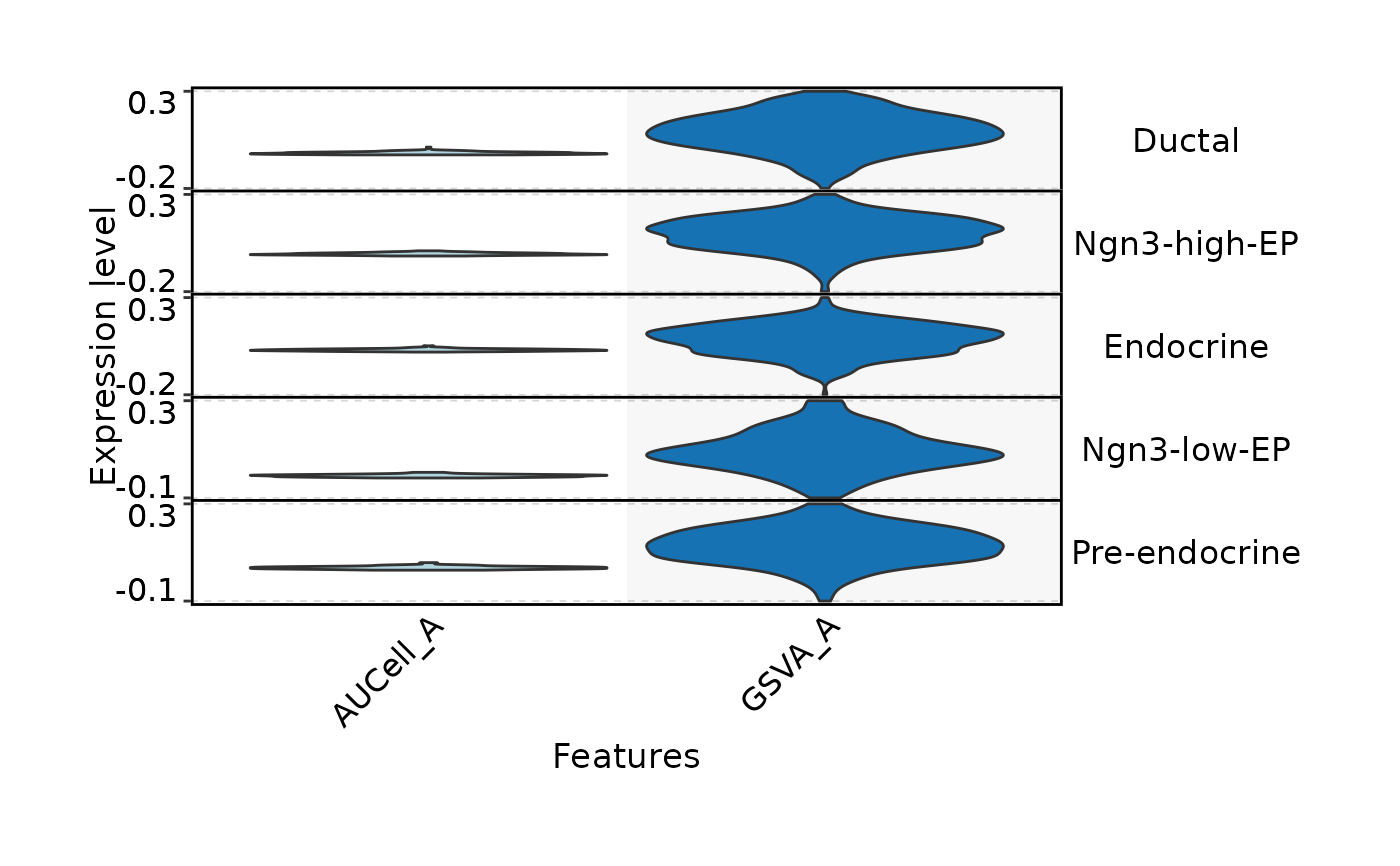 FeatureDimPlot(
pancreas_sub,
features = c("AUCell_A", "GSVA_A"),
xlab = "UMAP_1",
ylab = "UMAP_2"
)
FeatureDimPlot(
pancreas_sub,
features = c("AUCell_A", "GSVA_A"),
xlab = "UMAP_1",
ylab = "UMAP_2"
)
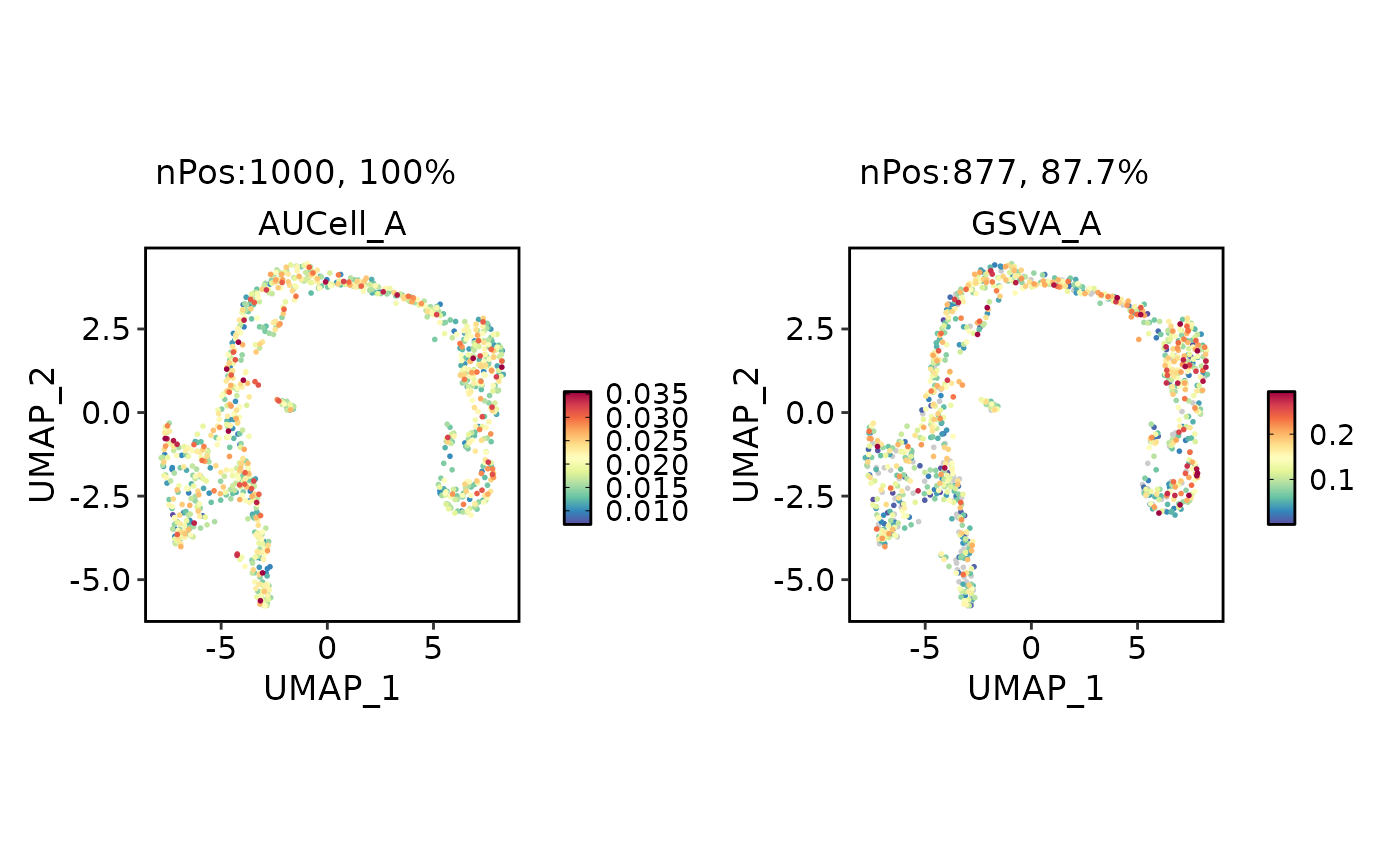 GroupHeatmap(
pancreas_sub,
features = c("AUCell_A", "GSVA_A", "Sox9", "Anxa2", "Bicc1"),
group.by = "CellType"
)
#> $plot
GroupHeatmap(
pancreas_sub,
features = c("AUCell_A", "GSVA_A", "Sox9", "Anxa2", "Bicc1"),
group.by = "CellType"
)
#> $plot
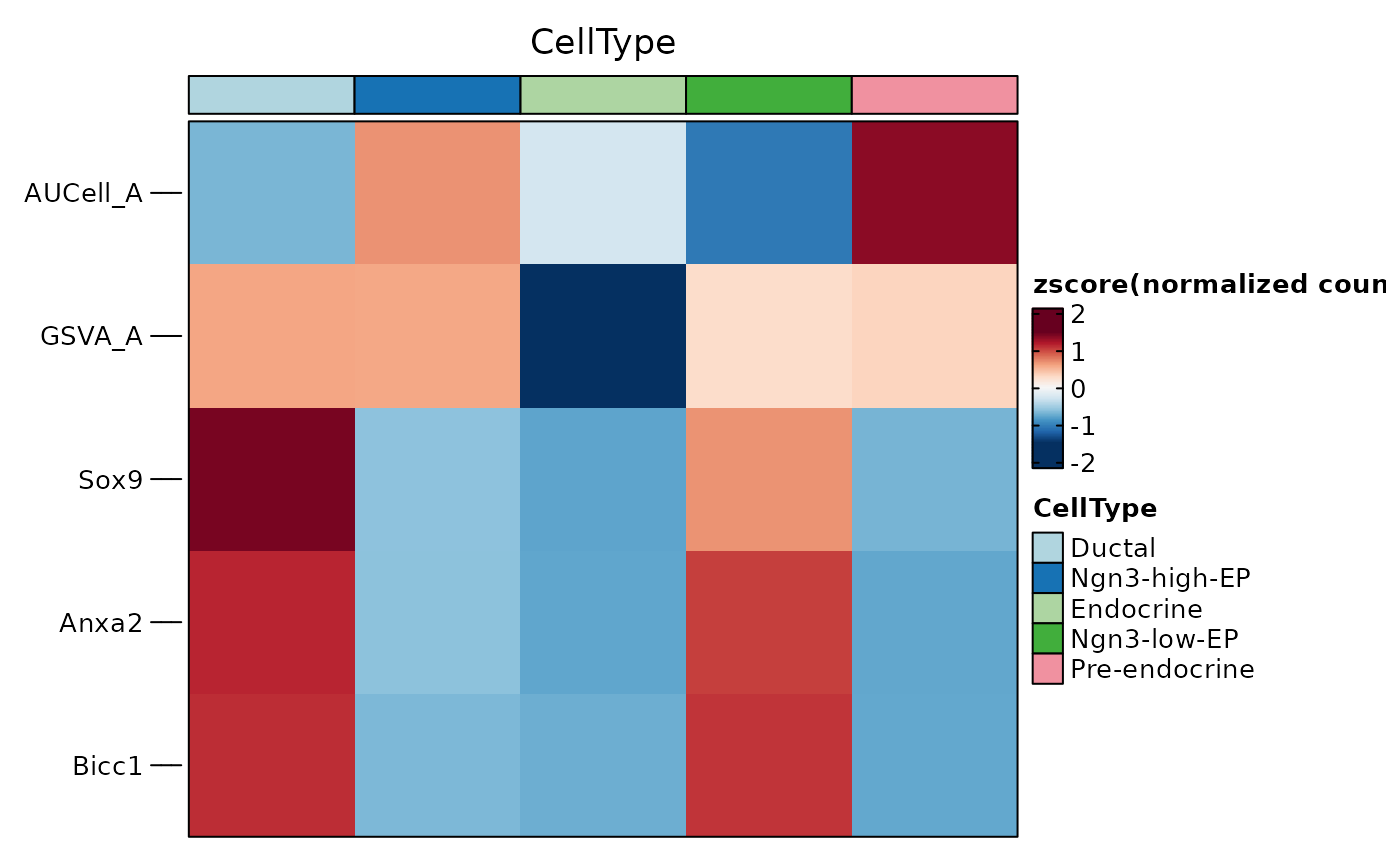 #>
#> $g_tree
#> gTree[GRID.gTree.11663]
#>
#> $matrix_list
#> $matrix_list$CellType
#> Ductal Ngn3-high-EP Endocrine Ngn3-low-EP Pre-endocrine
#> AUCell_A -0.6898028 0.6814256 -0.2740940 -1.0721911 1.3546623
#> GSVA_A 0.5904666 0.5803752 -1.7700722 0.2694525 0.3297779
#> Sox9 1.4288723 -0.6125486 -0.7946666 0.6788737 -0.7005309
#> Anxa2 1.1467252 -0.6170844 -0.7867363 1.0374551 -0.7803596
#> Bicc1 1.1109781 -0.6764747 -0.7375937 1.0784037 -0.7753133
#> attr(,"scaled:center")
#> AUCell_A GSVA_A Sox9 Anxa2 Bicc1
#> 0.01920529 0.10872510 1.96420268 2.04845477 1.94117464
#> attr(,"scaled:scale")
#> AUCell_A GSVA_A Sox9 Anxa2 Bicc1
#> 0.0007646939 0.0184930194 2.1633376549 2.5428690690 2.3797100097
#>
#>
#> $feature_split
#> NULL
#>
#> $cell_metadata
#> cells CellType
#> AAACCTGAGCCTTGAT AAACCTGAGCCTTGAT Ductal
#> AAACCTGGTAAGTGGC AAACCTGGTAAGTGGC Ngn3-high-EP
#> AAACGGGAGATATGGT AAACGGGAGATATGGT Ductal
#> AAACGGGCAAAGAATC AAACGGGCAAAGAATC Endocrine
#> AAACGGGGTACAGTTC AAACGGGGTACAGTTC Endocrine
#> AAACGGGTCAGCTCTC AAACGGGTCAGCTCTC Ngn3-low-EP
#> AAACGGGTCGCATGGC AAACGGGTCGCATGGC Endocrine
#> AAAGATGAGTCATGCT AAAGATGAGTCATGCT Endocrine
#> AAAGATGAGTCTTGCA AAAGATGAGTCTTGCA Endocrine
#> AAAGCAAAGCTACCTA AAAGCAAAGCTACCTA Pre-endocrine
#> AAAGCAACACATGGGA AAAGCAACACATGGGA Ductal
#> AAAGCAAGTGCAGTAG AAAGCAAGTGCAGTAG Endocrine
#> AAATGCCAGACTAAGT AAATGCCAGACTAAGT Endocrine
#> AAATGCCCAATGGAAT AAATGCCCAATGGAAT Endocrine
#> AAATGCCCACCAACCG AAATGCCCACCAACCG Ngn3-high-EP
#> AAATGCCCATGCTAGT AAATGCCCATGCTAGT Ngn3-high-EP
#> AAATGCCGTGATAAGT AAATGCCGTGATAAGT Endocrine
#> AACACGTGTCGACTGC AACACGTGTCGACTGC Pre-endocrine
#> AACCATGCACAGGAGT AACCATGCACAGGAGT Ngn3-high-EP
#> AACCATGGTTCGGCAC AACCATGGTTCGGCAC Ngn3-high-EP
#> AACCATGGTTGAACTC AACCATGGTTGAACTC Pre-endocrine
#> AACGTTGGTGCCTTGG AACGTTGGTGCCTTGG Ductal
#> AACTCAGGTGCCTGTG AACTCAGGTGCCTGTG Ngn3-high-EP
#> AACTCAGGTTGGAGGT AACTCAGGTTGGAGGT Ductal
#> AACTCAGTCCTCGCAT AACTCAGTCCTCGCAT Ngn3-high-EP
#> AACTCAGTCTGGTTCC AACTCAGTCTGGTTCC Ductal
#> AACTCCCCAGTTCATG AACTCCCCAGTTCATG Endocrine
#> AACTCCCTCGAGAGCA AACTCCCTCGAGAGCA Ductal
#> AACTCTTCATTTGCCC AACTCTTCATTTGCCC Endocrine
#> AACTGGTGTGCCTGTG AACTGGTGTGCCTGTG Ductal
#> AACTGGTTCTTACCGC AACTGGTTCTTACCGC Ductal
#> AACTTTCCACAGTCGC AACTTTCCACAGTCGC Ngn3-high-EP
#> AACTTTCCACTGCCAG AACTTTCCACTGCCAG Ductal
#> AACTTTCGTGACCAAG AACTTTCGTGACCAAG Ngn3-low-EP
#> AACTTTCTCCAGATCA AACTTTCTCCAGATCA Endocrine
#> AAGACCTAGACCTTTG AAGACCTAGACCTTTG Endocrine
#> AAGACCTAGTAGGCCA AAGACCTAGTAGGCCA Ngn3-high-EP
#> AAGACCTCAGGTTTCA AAGACCTCAGGTTTCA Ductal
#> AAGCCGCAGGCAGGTT AAGCCGCAGGCAGGTT Ductal
#> AAGCCGCCAATGGACG AAGCCGCCAATGGACG Ngn3-high-EP
#> AAGGAGCAGTACATGA AAGGAGCAGTACATGA Endocrine
#> AAGGCAGAGCTGAAAT AAGGCAGAGCTGAAAT Ductal
#> AAGGCAGTCCGAGCCA AAGGCAGTCCGAGCCA Ngn3-low-EP
#> AAGGTTCAGATAGTCA AAGGTTCAGATAGTCA Endocrine
#> AAGGTTCAGGAGCGAG AAGGTTCAGGAGCGAG Endocrine
#> AAGGTTCGTCAACTGT AAGGTTCGTCAACTGT Pre-endocrine
#> AAGTCTGGTTGGGACA AAGTCTGGTTGGGACA Endocrine
#> AAGTCTGTCCGAGCCA AAGTCTGTCCGAGCCA Ngn3-high-EP
#> AATCCAGAGGGTTCCC AATCCAGAGGGTTCCC Pre-endocrine
#> AATCCAGTCAGGTTCA AATCCAGTCAGGTTCA Pre-endocrine
#> AATCCAGTCGGTCCGA AATCCAGTCGGTCCGA Pre-endocrine
#> AATCGGTCATATGGTC AATCGGTCATATGGTC Ngn3-high-EP
#> AATCGGTTCCAAACTG AATCGGTTCCAAACTG Ngn3-high-EP
#> AATCGGTTCGTATCAG AATCGGTTCGTATCAG Endocrine
#> ACACCAAGTATAGGTA ACACCAAGTATAGGTA Endocrine
#> ACACCCTAGGACAGCT ACACCCTAGGACAGCT Ngn3-high-EP
#> ACACCCTTCCCAGGTG ACACCCTTCCCAGGTG Pre-endocrine
#> ACACCGGCATCACGTA ACACCGGCATCACGTA Ductal
#> ACACCGGCATGTCCTC ACACCGGCATGTCCTC Ductal
#> ACACCGGTCATGTCTT ACACCGGTCATGTCTT Endocrine
#> ACACTGAGTCCATGAT ACACTGAGTCCATGAT Pre-endocrine
#> ACAGCCGAGAGAACAG ACAGCCGAGAGAACAG Pre-endocrine
#> ACAGCCGAGAGGTACC ACAGCCGAGAGGTACC Endocrine
#> ACAGCCGAGTGGTCCC ACAGCCGAGTGGTCCC Ductal
#> ACAGCCGCATCGTCGG ACAGCCGCATCGTCGG Ngn3-high-EP
#> ACAGCTAAGTCGTTTG ACAGCTAAGTCGTTTG Endocrine
#> ACAGCTATCAAGGCTT ACAGCTATCAAGGCTT Endocrine
#> ACAGCTATCAGCATGT ACAGCTATCAGCATGT Ductal
#> ACAGCTATCTGGGCCA ACAGCTATCTGGGCCA Ductal
#> ACATACGAGTAGGCCA ACATACGAGTAGGCCA Ngn3-high-EP
#> ACATACGGTCTCTCTG ACATACGGTCTCTCTG Pre-endocrine
#> ACATCAGAGAGACTTA ACATCAGAGAGACTTA Pre-endocrine
#> ACATCAGTCCTTGGTC ACATCAGTCCTTGGTC Ngn3-high-EP
#> ACCAGTAAGTGATCGG ACCAGTAAGTGATCGG Endocrine
#> ACCAGTAGTTCGTTGA ACCAGTAGTTCGTTGA Endocrine
#> ACCAGTATCGTTTAGG ACCAGTATCGTTTAGG Ngn3-high-EP
#> ACCGTAAGTTATCACG ACCGTAAGTTATCACG Ductal
#> ACCTTTATCCTGCAGG ACCTTTATCCTGCAGG Endocrine
#> ACGAGCCCATGAGCGA ACGAGCCCATGAGCGA Endocrine
#> ACGAGGAAGTTACCCA ACGAGGAAGTTACCCA Ngn3-low-EP
#> ACGAGGATCATCACCC ACGAGGATCATCACCC Endocrine
#> ACGAGGATCGCCGTGA ACGAGGATCGCCGTGA Pre-endocrine
#> ACGATACAGATGAGAG ACGATACAGATGAGAG Endocrine
#> ACGATACTCGCGCCAA ACGATACTCGCGCCAA Endocrine
#> ACGATGTTCGCAGGCT ACGATGTTCGCAGGCT Pre-endocrine
#> ACGCAGCCATTCTCAT ACGCAGCCATTCTCAT Pre-endocrine
#> ACGCAGCGTAATCACC ACGCAGCGTAATCACC Ngn3-high-EP
#> ACGCCAGGTTACAGAA ACGCCAGGTTACAGAA Endocrine
#> ACGCCAGTCCCAAGTA ACGCCAGTCCCAAGTA Ngn3-high-EP
#> ACGCCGAAGGGTGTTG ACGCCGAAGGGTGTTG Endocrine
#> ACGCCGAGTTTGCATG ACGCCGAGTTTGCATG Endocrine
#> ACGGCCACACGTGAGA ACGGCCACACGTGAGA Ductal
#> ACGGGCTGTGCGATAG ACGGGCTGTGCGATAG Ductal
#> ACGTCAAAGGTGTTAA ACGTCAAAGGTGTTAA Endocrine
#> ACGTCAACACTGCCAG ACGTCAACACTGCCAG Ductal
#> ACGTCAACAGACAAAT ACGTCAACAGACAAAT Ductal
#> ACTATCTGTCAGGACA ACTATCTGTCAGGACA Endocrine
#> ACTATCTTCAAGATCC ACTATCTTCAAGATCC Pre-endocrine
#> ACTGAACAGCCTCGTG ACTGAACAGCCTCGTG Ductal
#> ACTGAGTAGTGACTCT ACTGAGTAGTGACTCT Ductal
#> ACTGATGCATCAGTAC ACTGATGCATCAGTAC Ngn3-high-EP
#> ACTGATGCATGGTCTA ACTGATGCATGGTCTA Endocrine
#> ACTGATGCATGTCTCC ACTGATGCATGTCTCC Pre-endocrine
#> ACTGATGGTGAGGGTT ACTGATGGTGAGGGTT Ductal
#> ACTGATGTCCCTAACC ACTGATGTCCCTAACC Ngn3-low-EP
#> ACTGCTCAGGTGTTAA ACTGCTCAGGTGTTAA Pre-endocrine
#> ACTGTCCCAGTTTACG ACTGTCCCAGTTTACG Pre-endocrine
#> ACTGTCCTCGCTAGCG ACTGTCCTCGCTAGCG Ngn3-high-EP
#> ACTTACTAGAGGGCTT ACTTACTAGAGGGCTT Ngn3-high-EP
#> ACTTACTAGCCGGTAA ACTTACTAGCCGGTAA Ngn3-low-EP
#> ACTTACTCAATCAGAA ACTTACTCAATCAGAA Ngn3-high-EP
#> ACTTACTCACATGTGT ACTTACTCACATGTGT Endocrine
#> ACTTGTTAGTGAAGTT ACTTGTTAGTGAAGTT Endocrine
#> ACTTGTTAGTTAAGTG ACTTGTTAGTTAAGTG Endocrine
#> ACTTGTTCATGGTCAT ACTTGTTCATGGTCAT Endocrine
#> ACTTGTTTCACCCGAG ACTTGTTTCACCCGAG Endocrine
#> ACTTTCAAGGATATAC ACTTTCAAGGATATAC Ductal
#> ACTTTCACAACTGCTA ACTTTCACAACTGCTA Ngn3-high-EP
#> AGAATAGCAATCACAC AGAATAGCAATCACAC Endocrine
#> AGAATAGCAGTACACT AGAATAGCAGTACACT Ductal
#> AGACGTTCAATGGTCT AGACGTTCAATGGTCT Endocrine
#> AGACGTTTCTTACCTA AGACGTTTCTTACCTA Ngn3-low-EP
#> AGAGCGACAGTAAGAT AGAGCGACAGTAAGAT Endocrine
#> AGAGCGATCTCTTGAT AGAGCGATCTCTTGAT Ductal
#> AGAGCTTAGCCGATTT AGAGCTTAGCCGATTT Ductal
#> AGAGCTTAGGTTACCT AGAGCTTAGGTTACCT Ngn3-high-EP
#> AGAGCTTGTTAAAGAC AGAGCTTGTTAAAGAC Ductal
#> AGAGTGGAGGCGATAC AGAGTGGAGGCGATAC Endocrine
#> AGAGTGGCACCTCGGA AGAGTGGCACCTCGGA Pre-endocrine
#> AGAGTGGGTAGCACGA AGAGTGGGTAGCACGA Ductal
#> AGAGTGGGTCACCTAA AGAGTGGGTCACCTAA Ngn3-low-EP
#> AGATCTGTCCGTCAAA AGATCTGTCCGTCAAA Pre-endocrine
#> AGATTGCAGCGTGTCC AGATTGCAGCGTGTCC Endocrine
#> AGATTGCAGCTGTCTA AGATTGCAGCTGTCTA Ductal
#> AGATTGCAGTTCCACA AGATTGCAGTTCCACA Ngn3-high-EP
#> AGATTGCCACCAGGCT AGATTGCCACCAGGCT Ductal
#> AGCAGCCGTCGCATCG AGCAGCCGTCGCATCG Ngn3-high-EP
#> AGCAGCCGTGTAACGG AGCAGCCGTGTAACGG Ngn3-high-EP
#> AGCAGCCTCTTTAGTC AGCAGCCTCTTTAGTC Ngn3-low-EP
#> AGCATACCACAGCCCA AGCATACCACAGCCCA Endocrine
#> AGCATACGTCCGCTGA AGCATACGTCCGCTGA Endocrine
#> AGCGGTCGTACCGAGA AGCGGTCGTACCGAGA Ngn3-high-EP
#> AGCGGTCGTCGAATCT AGCGGTCGTCGAATCT Ngn3-high-EP
#> AGCGGTCGTGTATGGG AGCGGTCGTGTATGGG Ductal
#> AGCGTCGCACAGGCCT AGCGTCGCACAGGCCT Ngn3-high-EP
#> AGCTCCTCAGTTTACG AGCTCCTCAGTTTACG Pre-endocrine
#> AGCTCCTGTGTGCCTG AGCTCCTGTGTGCCTG Ductal
#> AGCTCCTTCAATACCG AGCTCCTTCAATACCG Ngn3-high-EP
#> AGCTCCTTCGCCCTTA AGCTCCTTCGCCCTTA Pre-endocrine
#> AGCTCTCAGACATAAC AGCTCTCAGACATAAC Pre-endocrine
#> AGCTCTCGTTCAGTAC AGCTCTCGTTCAGTAC Endocrine
#> AGCTTGAAGACACGAC AGCTTGAAGACACGAC Ngn3-high-EP
#> AGCTTGAAGAGTGACC AGCTTGAAGAGTGACC Endocrine
#> AGCTTGACAATAGAGT AGCTTGACAATAGAGT Ngn3-low-EP
#> AGCTTGACAGTAAGAT AGCTTGACAGTAAGAT Ductal
#> AGCTTGACATCTATGG AGCTTGACATCTATGG Endocrine
#> AGCTTGAGTGCAGTAG AGCTTGAGTGCAGTAG Endocrine
#> AGGCCACAGCTGTCTA AGGCCACAGCTGTCTA Ngn3-low-EP
#> AGGCCACCATGACATC AGGCCACCATGACATC Endocrine
#> AGGCCGTCACGACGAA AGGCCGTCACGACGAA Ngn3-high-EP
#> AGGCCGTCACGGTGTC AGGCCGTCACGGTGTC Endocrine
#> AGGCCGTGTACCAGTT AGGCCGTGTACCAGTT Ngn3-low-EP
#> AGGGAGTAGGGTATCG AGGGAGTAGGGTATCG Ngn3-high-EP
#> AGGGAGTAGTGTCTCA AGGGAGTAGTGTCTCA Ductal
#> AGGGAGTTCCTCAATT AGGGAGTTCCTCAATT Endocrine
#> AGGGAGTTCGGAGGTA AGGGAGTTCGGAGGTA Endocrine
#> AGGGATGGTCGACTGC AGGGATGGTCGACTGC Endocrine
#> AGGGATGGTTAGGGTG AGGGATGGTTAGGGTG Ngn3-high-EP
#> AGGGATGTCGCATGAT AGGGATGTCGCATGAT Endocrine
#> AGGGATGTCGCATGGC AGGGATGTCGCATGGC Ngn3-high-EP
#> AGGGTGAGTGCGAAAC AGGGTGAGTGCGAAAC Endocrine
#> AGGTCATCACTTCTGC AGGTCATCACTTCTGC Endocrine
#> AGGTCATGTCTCCCTA AGGTCATGTCTCCCTA Endocrine
#> AGGTCATGTTTAGGAA AGGTCATGTTTAGGAA Pre-endocrine
#> AGTAGTCCACATGACT AGTAGTCCACATGACT Ngn3-low-EP
#> AGTAGTCGTGAGGGAG AGTAGTCGTGAGGGAG Endocrine
#> AGTAGTCGTTCTGTTT AGTAGTCGTTCTGTTT Ductal
#> AGTCTTTCACCAGGTC AGTCTTTCACCAGGTC Ductal
#> AGTCTTTCACGGACAA AGTCTTTCACGGACAA Endocrine
#> AGTCTTTGTTACGGAG AGTCTTTGTTACGGAG Endocrine
#> AGTGAGGGTTACGGAG AGTGAGGGTTACGGAG Ductal
#> AGTGAGGTCTCCAACC AGTGAGGTCTCCAACC Endocrine
#> AGTGGGACAGGTCGTC AGTGGGACAGGTCGTC Endocrine
#> AGTGGGAGTGGCAAAC AGTGGGAGTGGCAAAC Pre-endocrine
#> AGTGTCATCCCTTGTG AGTGTCATCCCTTGTG Pre-endocrine
#> AGTTGGTAGAGCCTAG AGTTGGTAGAGCCTAG Endocrine
#> AGTTGGTGTTGTCGCG AGTTGGTGTTGTCGCG Pre-endocrine
#> AGTTGGTTCACATAGC AGTTGGTTCACATAGC Ngn3-high-EP
#> ATAAGAGCATCGATTG ATAAGAGCATCGATTG Ductal
#> ATAGACCAGCGTGTCC ATAGACCAGCGTGTCC Endocrine
#> ATAGACCAGTGATCGG ATAGACCAGTGATCGG Endocrine
#> ATAGACCTCTAACTGG ATAGACCTCTAACTGG Ductal
#> ATCACGAGTGCACCAC ATCACGAGTGCACCAC Endocrine
#> ATCATCTGTGTTTGTG ATCATCTGTGTTTGTG Endocrine
#> ATCATCTTCTGTCAAG ATCATCTTCTGTCAAG Ductal
#> ATCATGGAGGAATGGA ATCATGGAGGAATGGA Endocrine
#> ATCATGGGTATTCGTG ATCATGGGTATTCGTG Endocrine
#> ATCATGGGTCCGCTGA ATCATGGGTCCGCTGA Ngn3-high-EP
#> ATCATGGTCCAGAGGA ATCATGGTCCAGAGGA Ductal
#> ATCCACCAGAGCCTAG ATCCACCAGAGCCTAG Endocrine
#> ATCCACCCAGCTCGCA ATCCACCCAGCTCGCA Endocrine
#> ATCCACCGTACCATCA ATCCACCGTACCATCA Ductal
#> ATCCGAAGTACACCGC ATCCGAAGTACACCGC Pre-endocrine
#> ATCCGAAGTGACTCAT ATCCGAAGTGACTCAT Ductal
#> ATCTGCCCAATCCGAT ATCTGCCCAATCCGAT Endocrine
#> ATCTGCCCATGTTGAC ATCTGCCCATGTTGAC Endocrine
#> ATGAGGGTCTTGACGA ATGAGGGTCTTGACGA Pre-endocrine
#> ATGTGTGCACAAGTAA ATGTGTGCACAAGTAA Ngn3-high-EP
#> ATGTGTGTCAAAGACA ATGTGTGTCAAAGACA Ngn3-high-EP
#> ATTACTCAGTTGCAGG ATTACTCAGTTGCAGG Ductal
#> ATTACTCTCAGGCGAA ATTACTCTCAGGCGAA Ductal
#> ATTACTCTCTGCCAGG ATTACTCTCTGCCAGG Endocrine
#> ATTATCCAGAAACCTA ATTATCCAGAAACCTA Ngn3-high-EP
#> ATTATCCAGACCCACC ATTATCCAGACCCACC Ngn3-high-EP
#> ATTATCCAGTGAACAT ATTATCCAGTGAACAT Ductal
#> ATTGGACAGTAATCCC ATTGGACAGTAATCCC Ngn3-low-EP
#> ATTGGTGTCCAGTATG ATTGGTGTCCAGTATG Ductal
#> ATTTCTGGTAGTGAAT ATTTCTGGTAGTGAAT Ngn3-low-EP
#> CAACCAAAGCGTTCCG CAACCAAAGCGTTCCG Pre-endocrine
#> CAACCAACAACCGCCA CAACCAACAACCGCCA Endocrine
#> CAACCAACACCTCGTT CAACCAACACCTCGTT Ngn3-high-EP
#> CAACCAATCGGGAGTA CAACCAATCGGGAGTA Ductal
#> CAAGAAAAGAGTACCG CAAGAAAAGAGTACCG Ngn3-high-EP
#> CAAGAAATCGTACCGG CAAGAAATCGTACCGG Ductal
#> CAAGGCCAGCTGTTCA CAAGGCCAGCTGTTCA Ductal
#> CAAGGCCAGTGTGGCA CAAGGCCAGTGTGGCA Ductal
#> CAAGGCCCACATTTCT CAAGGCCCACATTTCT Ngn3-high-EP
#> CAAGGCCTCAGTGCAT CAAGGCCTCAGTGCAT Endocrine
#> CAAGGCCTCTCAACTT CAAGGCCTCTCAACTT Pre-endocrine
#> CAAGGCCTCTTACCTA CAAGGCCTCTTACCTA Endocrine
#> CAAGTTGGTGTGCCTG CAAGTTGGTGTGCCTG Pre-endocrine
#> CAAGTTGTCTAACTGG CAAGTTGTCTAACTGG Pre-endocrine
#> CACAAACAGTAGCGGT CACAAACAGTAGCGGT Ngn3-high-EP
#> CACAAACCACGAAAGC CACAAACCACGAAAGC Endocrine
#> CACACAACAGCCAGAA CACACAACAGCCAGAA Ductal
#> CACACCTGTACATGTC CACACCTGTACATGTC Endocrine
#> CACACCTGTTATCCGA CACACCTGTTATCCGA Pre-endocrine
#> CACACTCAGATCCCAT CACACTCAGATCCCAT Ductal
#> CACACTCTCATGTGGT CACACTCTCATGTGGT Ngn3-low-EP
#> CACAGGCCAAGGCTCC CACAGGCCAAGGCTCC Pre-endocrine
#> CACAGGCCACGTCAGC CACAGGCCACGTCAGC Endocrine
#> CACAGTAGTGCACTTA CACAGTAGTGCACTTA Endocrine
#> CACAGTAGTTCCTCCA CACAGTAGTTCCTCCA Ductal
#> CACAGTATCCTCCTAG CACAGTATCCTCCTAG Ductal
#> CACATAGAGCGTGTCC CACATAGAGCGTGTCC Ductal
#> CACATTTGTTTCGCTC CACATTTGTTTCGCTC Ductal
#> CACATTTTCGAGGTAG CACATTTTCGAGGTAG Endocrine
#> CACATTTTCGCAGGCT CACATTTTCGCAGGCT Ductal
#> CACCACTAGCGATCCC CACCACTAGCGATCCC Endocrine
#> CACCACTGTGTGTGCC CACCACTGTGTGTGCC Pre-endocrine
#> CACCAGGCATAAAGGT CACCAGGCATAAAGGT Pre-endocrine
#> CACCTTGAGAGTAATC CACCTTGAGAGTAATC Ductal
#> CACTCCACATCGTCGG CACTCCACATCGTCGG Endocrine
#> CACTCCATCAACACAC CACTCCATCAACACAC Endocrine
#> CAGAATCCATTGGGCC CAGAATCCATTGGGCC Endocrine
#> CAGAATCGTCATTAGC CAGAATCGTCATTAGC Ngn3-high-EP
#> CAGAATCGTGTAAGTA CAGAATCGTGTAAGTA Ngn3-low-EP
#> CAGAGAGTCGGCCGAT CAGAGAGTCGGCCGAT Endocrine
#> CAGATCAAGCTGTTCA CAGATCAAGCTGTTCA Endocrine
#> CAGCAGCAGACAAAGG CAGCAGCAGACAAAGG Ductal
#> CAGCAGCCACACTGCG CAGCAGCCACACTGCG Ductal
#> CAGCAGCGTTCACGGC CAGCAGCGTTCACGGC Ductal
#> CAGCCGAAGCGATATA CAGCCGAAGCGATATA Ductal
#> CAGCCGAAGTAGGTGC CAGCCGAAGTAGGTGC Ductal
#> CAGCCGATCCTATTCA CAGCCGATCCTATTCA Ngn3-high-EP
#> CAGCCGATCGTTTATC CAGCCGATCGTTTATC Ngn3-high-EP
#> CAGCGACCACAGGAGT CAGCGACCACAGGAGT Ductal
#> CAGCTAAAGTCCATAC CAGCTAAAGTCCATAC Ductal
#> CAGCTAACATGCCTAA CAGCTAACATGCCTAA Pre-endocrine
#> CAGCTAAGTACCGAGA CAGCTAAGTACCGAGA Ngn3-high-EP
#> CAGCTGGCAAGTTGTC CAGCTGGCAAGTTGTC Pre-endocrine
#> CAGCTGGGTCTAACGT CAGCTGGGTCTAACGT Pre-endocrine
#> CAGCTGGGTTCTCATT CAGCTGGGTTCTCATT Endocrine
#> CAGGTGCAGCCAGGAT CAGGTGCAGCCAGGAT Endocrine
#> CAGGTGCCATCAGTCA CAGGTGCCATCAGTCA Pre-endocrine
#> CAGTCCTAGGTGGGTT CAGTCCTAGGTGGGTT Endocrine
#> CAGTCCTCAGGGCATA CAGTCCTCAGGGCATA Endocrine
#> CATATGGCATGTTGAC CATATGGCATGTTGAC Ductal
#> CATATGGGTTACTGAC CATATGGGTTACTGAC Endocrine
#> CATATGGTCACAGTAC CATATGGTCACAGTAC Pre-endocrine
#> CATATTCTCCTGTAGA CATATTCTCCTGTAGA Endocrine
#> CATCAGACATGCATGT CATCAGACATGCATGT Ductal
#> CATCAGAGTCTCTTTA CATCAGAGTCTCTTTA Pre-endocrine
#> CATCAGATCATCGCTC CATCAGATCATCGCTC Pre-endocrine
#> CATCGAAGTACGAAAT CATCGAAGTACGAAAT Endocrine
#> CATCGAAGTCACACGC CATCGAAGTCACACGC Endocrine
#> CATCGGGCACGTTGGC CATCGGGCACGTTGGC Endocrine
#> CATCGGGTCTTCGGTC CATCGGGTCTTCGGTC Ngn3-high-EP
#> CATGACACAACAACCT CATGACACAACAACCT Ngn3-low-EP
#> CATGACACATCGATTG CATGACACATCGATTG Ngn3-high-EP
#> CATGACAGTAGCTTGT CATGACAGTAGCTTGT Ngn3-low-EP
#> CATGACATCATGCATG CATGACATCATGCATG Endocrine
#> CATGCCTCACCCATTC CATGCCTCACCCATTC Endocrine
#> CATGGCGAGAGTCTGG CATGGCGAGAGTCTGG Ngn3-high-EP
#> CATTATCGTCATATCG CATTATCGTCATATCG Pre-endocrine
#> CCAATCCAGAGTAAGG CCAATCCAGAGTAAGG Ductal
#> CCACCTAAGACTTGAA CCACCTAAGACTTGAA Endocrine
#> CCACCTACAGCTCGAC CCACCTACAGCTCGAC Endocrine
#> CCACCTATCACCAGGC CCACCTATCACCAGGC Ductal
#> CCACCTATCTAAGCCA CCACCTATCTAAGCCA Endocrine
#> CCACGGAAGATCCTGT CCACGGAAGATCCTGT Endocrine
#> CCACGGAAGCAATCTC CCACGGAAGCAATCTC Endocrine
#> CCACGGAAGGGTGTTG CCACGGAAGGGTGTTG Endocrine
#> CCACGGAAGTCTCCTC CCACGGAAGTCTCCTC Pre-endocrine
#> CCACGGAGTCAACATC CCACGGAGTCAACATC Ngn3-high-EP
#> CCACGGAGTCTAGTCA CCACGGAGTCTAGTCA Endocrine
#> CCACGGAGTTACGACT CCACGGAGTTACGACT Ngn3-high-EP
#> CCACTACAGAGCTGGT CCACTACAGAGCTGGT Pre-endocrine
#> CCACTACAGAGTACCG CCACTACAGAGTACCG Ngn3-low-EP
#> CCACTACCAACGCACC CCACTACCAACGCACC Endocrine
#> CCAGCGACAGACGTAG CCAGCGACAGACGTAG Pre-endocrine
#> CCATGTCGTCGGCTCA CCATGTCGTCGGCTCA Ngn3-high-EP
#> CCATGTCTCCGCATAA CCATGTCTCCGCATAA Ngn3-high-EP
#> CCATTCGCATGCCTAA CCATTCGCATGCCTAA Ductal
#> CCATTCGGTAGGGACT CCATTCGGTAGGGACT Ductal
#> CCCAATCCAACTGGCC CCCAATCCAACTGGCC Endocrine
#> CCCAGTTAGCATCATC CCCAGTTAGCATCATC Endocrine
#> CCCAGTTGTCGTGGCT CCCAGTTGTCGTGGCT Endocrine
#> CCCAGTTGTGCGCTTG CCCAGTTGTGCGCTTG Endocrine
#> CCCAGTTTCAGCACAT CCCAGTTTCAGCACAT Endocrine
#> CCCATACAGACTTTCG CCCATACAGACTTTCG Endocrine
#> CCCATACCACATCCAA CCCATACCACATCCAA Ductal
#> CCCATACCATGCAACT CCCATACCATGCAACT Endocrine
#> CCCATACTCGTGGACC CCCATACTCGTGGACC Pre-endocrine
#> CCGGGATAGATCTGAA CCGGGATAGATCTGAA Ngn3-high-EP
#> CCGGGATTCGACAGCC CCGGGATTCGACAGCC Endocrine
#> CCGGTAGTCGGCTTGG CCGGTAGTCGGCTTGG Endocrine
#> CCGTACTAGGAGTACC CCGTACTAGGAGTACC Ngn3-high-EP
#> CCGTACTAGTCCGTAT CCGTACTAGTCCGTAT Ngn3-high-EP
#> CCGTACTTCAATAAGG CCGTACTTCAATAAGG Ngn3-low-EP
#> CCGTGGACATAGAAAC CCGTGGACATAGAAAC Endocrine
#> CCGTGGATCCAAGCCG CCGTGGATCCAAGCCG Ductal
#> CCGTGGATCTGTCTCG CCGTGGATCTGTCTCG Endocrine
#> CCGTGGATCTTAGAGC CCGTGGATCTTAGAGC Ngn3-high-EP
#> CCGTTCAAGCTAAACA CCGTTCAAGCTAAACA Endocrine
#> CCGTTCAAGGACCACA CCGTTCAAGGACCACA Ngn3-high-EP
#> CCGTTCACACATGACT CCGTTCACACATGACT Ngn3-low-EP
#> CCTACACAGATCCGAG CCTACACAGATCCGAG Ngn3-low-EP
#> CCTACACAGTACGTTC CCTACACAGTACGTTC Endocrine
#> CCTACACTCCTTTACA CCTACACTCCTTTACA Ductal
#> CCTACACTCTTTACAC CCTACACTCTTTACAC Endocrine
#> CCTACCACATCGATTG CCTACCACATCGATTG Ngn3-high-EP
#> CCTACCAGTTGCTCCT CCTACCAGTTGCTCCT Pre-endocrine
#> CCTACCATCTGCCCTA CCTACCATCTGCCCTA Endocrine
#> CCTAGCTCAGACTCGC CCTAGCTCAGACTCGC Ductal
#> CCTAGCTCATACTACG CCTAGCTCATACTACG Ductal
#> CCTAGCTGTGGCCCTA CCTAGCTGTGGCCCTA Endocrine
#> CCTAGCTTCGATGAGG CCTAGCTTCGATGAGG Ductal
#> CCTATTACAGACAAGC CCTATTACAGACAAGC Endocrine
#> CCTATTATCATCGCTC CCTATTATCATCGCTC Ngn3-high-EP
#> CCTCAGTAGTGCGATG CCTCAGTAGTGCGATG Pre-endocrine
#> CCTCAGTCAGACACTT CCTCAGTCAGACACTT Endocrine
#> CCTCAGTGTTACCAGT CCTCAGTGTTACCAGT Ngn3-high-EP
#> CCTCTGAGTATAAACG CCTCTGAGTATAAACG Ductal
#> CCTTACGAGATCACGG CCTTACGAGATCACGG Ductal
#> CCTTACGCACAACGCC CCTTACGCACAACGCC Pre-endocrine
#> CCTTCCCAGGCAGGTT CCTTCCCAGGCAGGTT Ductal
#> CCTTCCCCAAAGTGCG CCTTCCCCAAAGTGCG Ductal
#> CCTTCCCTCGTCGTTC CCTTCCCTCGTCGTTC Pre-endocrine
#> CCTTCGAAGATAGCAT CCTTCGAAGATAGCAT Endocrine
#> CCTTCGACAGCTGTTA CCTTCGACAGCTGTTA Ngn3-low-EP
#> CCTTCGATCACAGGCC CCTTCGATCACAGGCC Endocrine
#> CGAACATAGCCAGTAG CGAACATAGCCAGTAG Ductal
#> CGAACATCAGACGTAG CGAACATCAGACGTAG Ductal
#> CGAATGTAGTTCCACA CGAATGTAGTTCCACA Ductal
#> CGAATGTGTCTGGAGA CGAATGTGTCTGGAGA Pre-endocrine
#> CGAATGTTCACCGGGT CGAATGTTCACCGGGT Pre-endocrine
#> CGAATGTTCTTCATGT CGAATGTTCTTCATGT Endocrine
#> CGACCTTAGCGCTTAT CGACCTTAGCGCTTAT Ngn3-low-EP
#> CGACCTTAGGCTAGAC CGACCTTAGGCTAGAC Endocrine
#> CGACTTCCAGCCTATA CGACTTCCAGCCTATA Ductal
#> CGACTTCCAGCTGCAC CGACTTCCAGCTGCAC Pre-endocrine
#> CGACTTCTCACGCGGT CGACTTCTCACGCGGT Endocrine
#> CGAGAAGAGGGTTCCC CGAGAAGAGGGTTCCC Endocrine
#> CGAGAAGCACGCATCG CGAGAAGCACGCATCG Ngn3-high-EP
#> CGAGAAGTCGTTACAG CGAGAAGTCGTTACAG Endocrine
#> CGAGCACAGCTGAAAT CGAGCACAGCTGAAAT Ductal
#> CGAGCACAGGCAGGTT CGAGCACAGGCAGGTT Pre-endocrine
#> CGAGCACGTAGTAGTA CGAGCACGTAGTAGTA Pre-endocrine
#> CGAGCACTCTACCTGC CGAGCACTCTACCTGC Ductal
#> CGAGCACTCTTCGAGA CGAGCACTCTTCGAGA Ngn3-low-EP
#> CGATCGGAGCTGCAAG CGATCGGAGCTGCAAG Pre-endocrine
#> CGATCGGCACGCCAGT CGATCGGCACGCCAGT Ngn3-high-EP
#> CGATGGCGTTACGGAG CGATGGCGTTACGGAG Ductal
#> CGATGGCTCAGTGTTG CGATGGCTCAGTGTTG Endocrine
#> CGATGGCTCTGTGCAA CGATGGCTCTGTGCAA Ductal
#> CGATGTAAGTTTAGGA CGATGTAAGTTTAGGA Endocrine
#> CGATGTAGTACTCTCC CGATGTAGTACTCTCC Ngn3-high-EP
#> CGATTGAAGTCAAGGC CGATTGAAGTCAAGGC Pre-endocrine
#> CGCCAAGCAAAGTCAA CGCCAAGCAAAGTCAA Ngn3-high-EP
#> CGCCAAGCACCGGAAA CGCCAAGCACCGGAAA Ductal
#> CGCCAAGTCTAAGCCA CGCCAAGTCTAAGCCA Endocrine
#> CGCGGTAAGAGCTGGT CGCGGTAAGAGCTGGT Endocrine
#> CGCGGTAAGCTGCAAG CGCGGTAAGCTGCAAG Endocrine
#> CGCGTTTCAGTCTTCC CGCGTTTCAGTCTTCC Endocrine
#> CGCTGGAGTCCATCCT CGCTGGAGTCCATCCT Ductal
#> CGCTTCATCTCTGTCG CGCTTCATCTCTGTCG Endocrine
#> CGGACACAGTGTCTCA CGGACACAGTGTCTCA Ductal
#> CGGACGTAGACACGAC CGGACGTAGACACGAC Endocrine
#> CGGACTGAGGTAGCTG CGGACTGAGGTAGCTG Ductal
#> CGGACTGTCTTGAGAC CGGACTGTCTTGAGAC Endocrine
#> CGGAGCTAGATAGTCA CGGAGCTAGATAGTCA Endocrine
#> CGGAGCTCATTGGGCC CGGAGCTCATTGGGCC Ductal
#> CGGAGTCAGTTTAGGA CGGAGTCAGTTTAGGA Endocrine
#> CGGCTAGGTACTCGCG CGGCTAGGTACTCGCG Ngn3-high-EP
#> CGGGTCAAGGCGATAC CGGGTCAAGGCGATAC Endocrine
#> CGGGTCAAGGCTCAGA CGGGTCAAGGCTCAGA Ductal
#> CGGTTAAAGAGCTGCA CGGTTAAAGAGCTGCA Ductal
#> CGGTTAAGTAAACACA CGGTTAAGTAAACACA Ngn3-low-EP
#> CGGTTAAGTCTAGCCG CGGTTAAGTCTAGCCG Ngn3-high-EP
#> CGGTTAATCACCGGGT CGGTTAATCACCGGGT Pre-endocrine
#> CGGTTAATCGTCTGAA CGGTTAATCGTCTGAA Ductal
#> CGTAGCGTCGGAAATA CGTAGCGTCGGAAATA Pre-endocrine
#> CGTAGGCAGCGTAGTG CGTAGGCAGCGTAGTG Ductal
#> CGTCACTAGGGTGTGT CGTCACTAGGGTGTGT Ductal
#> CGTCACTCACGAAATA CGTCACTCACGAAATA Ngn3-low-EP
#> CGTCACTCATAGTAAG CGTCACTCATAGTAAG Ngn3-low-EP
#> CGTCACTTCACTTATC CGTCACTTCACTTATC Pre-endocrine
#> CGTCACTTCTAACTCT CGTCACTTCTAACTCT Endocrine
#> CGTCAGGAGCCTATGT CGTCAGGAGCCTATGT Ngn3-low-EP
#> CGTCAGGCATGGGAAC CGTCAGGCATGGGAAC Ngn3-low-EP
#> CGTCAGGGTAGGCATG CGTCAGGGTAGGCATG Ngn3-high-EP
#> CGTCAGGTCCGCAAGC CGTCAGGTCCGCAAGC Endocrine
#> CGTCAGGTCCTGCCAT CGTCAGGTCCTGCCAT Ductal
#> CGTCCATGTCTAACGT CGTCCATGTCTAACGT Endocrine
#> CGTCCATTCGCGCCAA CGTCCATTCGCGCCAA Ngn3-high-EP
#> CGTCTACAGATCTGCT CGTCTACAGATCTGCT Ductal
#> CGTCTACAGGGAGTAA CGTCTACAGGGAGTAA Endocrine
#> CGTGAGCTCAGCGATT CGTGAGCTCAGCGATT Ngn3-high-EP
#> CGTGAGCTCTTCTGGC CGTGAGCTCTTCTGGC Endocrine
#> CGTGTAAGTCTTGATG CGTGTAAGTCTTGATG Ngn3-low-EP
#> CGTGTAATCATTATCC CGTGTAATCATTATCC Ngn3-high-EP
#> CGTGTAATCCATGCTC CGTGTAATCCATGCTC Ductal
#> CGTGTCTCAGAAGCAC CGTGTCTCAGAAGCAC Endocrine
#> CGTGTCTGTAGCGTCC CGTGTCTGTAGCGTCC Endocrine
#> CGTTAGAAGTATCTCG CGTTAGAAGTATCTCG Ductal
#> CGTTAGAGTGCAACGA CGTTAGAGTGCAACGA Ductal
#> CGTTCTGAGGACGAAA CGTTCTGAGGACGAAA Endocrine
#> CGTTCTGTCACAATGC CGTTCTGTCACAATGC Ngn3-high-EP
#> CGTTCTGTCGATAGAA CGTTCTGTCGATAGAA Endocrine
#> CGTTGGGCAGGCGATA CGTTGGGCAGGCGATA Pre-endocrine
#> CTAACTTAGAGGTACC CTAACTTAGAGGTACC Endocrine
#> CTAACTTAGATCACGG CTAACTTAGATCACGG Endocrine
#> CTAACTTCATCTGGTA CTAACTTCATCTGGTA Endocrine
#> CTAACTTCATTGGGCC CTAACTTCATTGGGCC Ductal
#> CTAAGACAGCACGCCT CTAAGACAGCACGCCT Ngn3-high-EP
#> CTAAGACAGCCACGCT CTAAGACAGCCACGCT Ngn3-low-EP
#> CTAAGACGTATCAGTC CTAAGACGTATCAGTC Endocrine
#> CTAAGACTCTGCGGCA CTAAGACTCTGCGGCA Endocrine
#> CTAATGGCATGGTTGT CTAATGGCATGGTTGT Pre-endocrine
#> CTACACCCATCAGTCA CTACACCCATCAGTCA Endocrine
#> CTACACCGTCGAATCT CTACACCGTCGAATCT Ductal
#> CTACACCGTCGCCATG CTACACCGTCGCCATG Ductal
#> CTACATTCACCGTTGG CTACATTCACCGTTGG Pre-endocrine
#> CTACATTGTTACTGAC CTACATTGTTACTGAC Pre-endocrine
#> CTACCCAGTGCGCTTG CTACCCAGTGCGCTTG Ductal
#> CTACGTCAGCTAGGCA CTACGTCAGCTAGGCA Pre-endocrine
#> CTAGAGTCACTGTTAG CTAGAGTCACTGTTAG Ductal
#> CTAGCCTGTCTCTTAT CTAGCCTGTCTCTTAT Ductal
#> CTAGCCTGTTCGGGCT CTAGCCTGTTCGGGCT Ductal
#> CTAGCCTTCAGCGATT CTAGCCTTCAGCGATT Ductal
#> CTAGTGATCAAAGACA CTAGTGATCAAAGACA Endocrine
#> CTAGTGATCCCAGGTG CTAGTGATCCCAGGTG Endocrine
#> CTCACACGTCACACGC CTCACACGTCACACGC Ngn3-high-EP
#> CTCAGAACACTAGTAC CTCAGAACACTAGTAC Ngn3-high-EP
#> CTCAGAACATAGGATA CTCAGAACATAGGATA Endocrine
#> CTCAGAAGTCCTCTTG CTCAGAAGTCCTCTTG Ductal
#> CTCAGAATCAGGTTCA CTCAGAATCAGGTTCA Ngn3-low-EP
#> CTCATTACAAGCGCTC CTCATTACAAGCGCTC Ductal
#> CTCCTAGCACAACGTT CTCCTAGCACAACGTT Pre-endocrine
#> CTCGAAAGTAAGAGGA CTCGAAAGTAAGAGGA Ngn3-high-EP
#> CTCGAAAGTGGTGTAG CTCGAAAGTGGTGTAG Endocrine
#> CTCGAAATCCTGCCAT CTCGAAATCCTGCCAT Endocrine
#> CTCGAGGCAAGTCTGT CTCGAGGCAAGTCTGT Endocrine
#> CTCGAGGGTAAGTAGT CTCGAGGGTAAGTAGT Endocrine
#> CTCGAGGTCTGTTGAG CTCGAGGTCTGTTGAG Ngn3-high-EP
#> CTCGGAGGTCATTAGC CTCGGAGGTCATTAGC Pre-endocrine
#> CTCGGAGGTGAAGGCT CTCGGAGGTGAAGGCT Pre-endocrine
#> CTCGTACCAATGGATA CTCGTACCAATGGATA Ductal
#> CTCGTACTCTTTAGTC CTCGTACTCTTTAGTC Endocrine
#> CTCGTCAAGAACAATC CTCGTCAAGAACAATC Endocrine
#> CTCGTCACAACTGCTA CTCGTCACAACTGCTA Ductal
#> CTCGTCATCGGTCTAA CTCGTCATCGGTCTAA Ductal
#> CTCTAATAGTTATCGC CTCTAATAGTTATCGC Endocrine
#> CTCTGGTAGCAATCTC CTCTGGTAGCAATCTC Ngn3-high-EP
#> CTCTGGTAGTCCCACG CTCTGGTAGTCCCACG Ductal
#> CTCTGGTCAACACCTA CTCTGGTCAACACCTA Ductal
#> CTCTGGTCAAGACACG CTCTGGTCAAGACACG Endocrine
#> CTCTGGTCAAGTTGTC CTCTGGTCAAGTTGTC Pre-endocrine
#> CTCTGGTCAGTTCATG CTCTGGTCAGTTCATG Ngn3-low-EP
#> CTGAAACAGACTAGGC CTGAAACAGACTAGGC Ductal
#> CTGAAACGTAGCGCAA CTGAAACGTAGCGCAA Endocrine
#> CTGAAACGTATGGTTC CTGAAACGTATGGTTC Endocrine
#> CTGAAACTCGATAGAA CTGAAACTCGATAGAA Ngn3-high-EP
#> CTGAAACTCGCCGTGA CTGAAACTCGCCGTGA Ductal
#> CTGAAACTCTGTTTGT CTGAAACTCTGTTTGT Ngn3-high-EP
#> CTGAAGTGTTTGGGCC CTGAAGTGTTTGGGCC Endocrine
#> CTGAAGTTCTGCGGCA CTGAAGTTCTGCGGCA Pre-endocrine
#> CTGATCCTCAGTTGAC CTGATCCTCAGTTGAC Ductal
#> CTGCCTAGTCTCCATC CTGCCTAGTCTCCATC Ductal
#> CTGCGGACAGACTCGC CTGCGGACAGACTCGC Ngn3-high-EP
#> CTGCGGACATTAGCCA CTGCGGACATTAGCCA Pre-endocrine
#> CTGCGGATCAGCATGT CTGCGGATCAGCATGT Ductal
#> CTGCTGTAGACTGGGT CTGCTGTAGACTGGGT Pre-endocrine
#> CTGCTGTCAAAGCAAT CTGCTGTCAAAGCAAT Endocrine
#> CTGGTCTAGGATTCGG CTGGTCTAGGATTCGG Pre-endocrine
#> CTGGTCTAGGTGATTA CTGGTCTAGGTGATTA Endocrine
#> CTGGTCTCAGGAATGC CTGGTCTCAGGAATGC Ductal
#> CTGGTCTTCGCATGGC CTGGTCTTCGCATGGC Ngn3-high-EP
#> CTGTGCTCATTACCTT CTGTGCTCATTACCTT Ngn3-high-EP
#> CTTAACTCAAGCGTAG CTTAACTCAAGCGTAG Ductal
#> CTTAACTTCAGCTTAG CTTAACTTCAGCTTAG Pre-endocrine
#> CTTAGGACACAGGTTT CTTAGGACACAGGTTT Ductal
#> CTTAGGAGTCCGAATT CTTAGGAGTCCGAATT Ductal
#> CTTAGGATCAACGGGA CTTAGGATCAACGGGA Endocrine
#> CTTAGGATCACTCTTA CTTAGGATCACTCTTA Ngn3-high-EP
#> CTTCTCTAGGCAGGTT CTTCTCTAGGCAGGTT Ductal
#> CTTCTCTAGTCCGTAT CTTCTCTAGTCCGTAT Pre-endocrine
#> CTTCTCTCAGCATACT CTTCTCTCAGCATACT Endocrine
#> CTTCTCTGTCAGAGGT CTTCTCTGTCAGAGGT Endocrine
#> CTTGGCTAGACAGACC CTTGGCTAGACAGACC Ductal
#> CTTGGCTAGGACACCA CTTGGCTAGGACACCA Ductal
#> CTTGGCTCAAAGGAAG CTTGGCTCAAAGGAAG Pre-endocrine
#> CTTGGCTCAGCCACCA CTTGGCTCAGCCACCA Ngn3-high-EP
#> CTTGGCTCATCCCACT CTTGGCTCATCCCACT Endocrine
#> CTTGGCTGTTAGAACA CTTGGCTGTTAGAACA Endocrine
#> CTTTGCGAGACCACGA CTTTGCGAGACCACGA Pre-endocrine
#> CTTTGCGAGGAATGGA CTTTGCGAGGAATGGA Ngn3-low-EP
#> CTTTGCGTCCACGTGG CTTTGCGTCCACGTGG Endocrine
#> GAAACTCAGAAACCTA GAAACTCAGAAACCTA Endocrine
#> GAAATGAAGCTCCTCT GAAATGAAGCTCCTCT Ngn3-high-EP
#> GAAATGACAACTGGCC GAAATGACAACTGGCC Ngn3-low-EP
#> GAAATGACACATGACT GAAATGACACATGACT Ductal
#> GAAATGAGTCACCCAG GAAATGAGTCACCCAG Ngn3-high-EP
#> GAAATGAGTCGGATCC GAAATGAGTCGGATCC Ductal
#> GAACATCGTAATCGTC GAACATCGTAATCGTC Ductal
#> GAACCTAAGTAGATGT GAACCTAAGTAGATGT Endocrine
#> GAACCTATCACTTATC GAACCTATCACTTATC Endocrine
#> GAACGGAAGTCAAGCG GAACGGAAGTCAAGCG Endocrine
#> GAACGGACAAGGACAC GAACGGACAAGGACAC Endocrine
#> GAAGCAGGTCGCGAAA GAAGCAGGTCGCGAAA Pre-endocrine
#> GAAGCAGGTCTAGAGG GAAGCAGGTCTAGAGG Endocrine
#> GAAGCAGTCAAGGCTT GAAGCAGTCAAGGCTT Ductal
#> GAATGAAAGAATGTGT GAATGAAAGAATGTGT Ngn3-high-EP
#> GAATGAACAATCGAAA GAATGAACAATCGAAA Ductal
#> GAATGAACAGCCTTGG GAATGAACAGCCTTGG Ductal
#> GAATGAATCATCGGAT GAATGAATCATCGGAT Endocrine
#> GACACGCCAAAGGAAG GACACGCCAAAGGAAG Ductal
#> GACCAATAGAATGTGT GACCAATAGAATGTGT Endocrine
#> GACCAATCAGACGCCT GACCAATCAGACGCCT Pre-endocrine
#> GACCAATGTTATCCGA GACCAATGTTATCCGA Endocrine
#> GACCAATGTTGGACCC GACCAATGTTGGACCC Endocrine
#> GACCAATTCAACGGCC GACCAATTCAACGGCC Ngn3-high-EP
#> GACCTGGGTTCGTGAT GACCTGGGTTCGTGAT Endocrine
#> GACGCGTCAGTCGTGC GACGCGTCAGTCGTGC Ductal
#> GACGCGTGTCGGCTCA GACGCGTGTCGGCTCA Endocrine
#> GACGGCTAGGTGATAT GACGGCTAGGTGATAT Ductal
#> GACGGCTTCAACTCTT GACGGCTTCAACTCTT Pre-endocrine
#> GACGTGCGTCACCTAA GACGTGCGTCACCTAA Pre-endocrine
#> GACGTGCTCCGGCACA GACGTGCTCCGGCACA Endocrine
#> GACGTTAAGACGCAAC GACGTTAAGACGCAAC Ductal
#> GACGTTAAGACGCTTT GACGTTAAGACGCTTT Ngn3-high-EP
#> GACGTTACATCTCGCT GACGTTACATCTCGCT Endocrine
#> GACGTTAGTGTGAATA GACGTTAGTGTGAATA Pre-endocrine
#> GACTAACGTCATCCCT GACTAACGTCATCCCT Ngn3-high-EP
#> GACTAACTCAGTCCCT GACTAACTCAGTCCCT Ductal
#> GACTACAGTTCGTGAT GACTACAGTTCGTGAT Ductal
#> GACTGCGAGAATCTCC GACTGCGAGAATCTCC Ngn3-high-EP
#> GAGCAGAAGCGTGTCC GAGCAGAAGCGTGTCC Ductal
#> GAGCAGAGTTTGCATG GAGCAGAGTTTGCATG Endocrine
#> GAGCAGATCAACTCTT GAGCAGATCAACTCTT Pre-endocrine
#> GAGGTGAGTCCGCTGA GAGGTGAGTCCGCTGA Pre-endocrine
#> GAGGTGAGTGACAAAT GAGGTGAGTGACAAAT Ductal
#> GAGTCCGAGCGATGAC GAGTCCGAGCGATGAC Ngn3-high-EP
#> GAGTCCGAGTTAAGTG GAGTCCGAGTTAAGTG Ductal
#> GAGTCCGCAATTGCTG GAGTCCGCAATTGCTG Ductal
#> GATCAGTAGGTGATAT GATCAGTAGGTGATAT Endocrine
#> GATCGATAGAAAGTGG GATCGATAGAAAGTGG Pre-endocrine
#> GATCGATAGGATGGTC GATCGATAGGATGGTC Endocrine
#> GATCGATGTAGCGATG GATCGATGTAGCGATG Endocrine
#> GATCGATGTTGTTTGG GATCGATGTTGTTTGG Endocrine
#> GATCGCGCACCAGGCT GATCGCGCACCAGGCT Pre-endocrine
#> GATCGTAAGTAGGTGC GATCGTAAGTAGGTGC Pre-endocrine
#> GATCGTAGTCCTAGCG GATCGTAGTCCTAGCG Ductal
#> GATCTAGAGCTAGCCC GATCTAGAGCTAGCCC Endocrine
#> GATCTAGGTGCCTTGG GATCTAGGTGCCTTGG Ductal
#> GATGAAAAGTTGTAGA GATGAAAAGTTGTAGA Ngn3-low-EP
#> GATGAAACAGGCTGAA GATGAAACAGGCTGAA Endocrine
#> GATGAAACATGGTTGT GATGAAACATGGTTGT Ductal
#> GATGAGGAGTGGACGT GATGAGGAGTGGACGT Ngn3-high-EP
#> GATGAGGTCTGGGCCA GATGAGGTCTGGGCCA Ductal
#> GATGCTAGTAGAAGGA GATGCTAGTAGAAGGA Ductal
#> GATGCTAGTGTTAAGA GATGCTAGTGTTAAGA Pre-endocrine
#> GATTCAGAGCGATGAC GATTCAGAGCGATGAC Pre-endocrine
#> GATTCAGAGTACGTTC GATTCAGAGTACGTTC Endocrine
#> GATTCAGTCGCCAGCA GATTCAGTCGCCAGCA Pre-endocrine
#> GATTCAGTCGTCCAGG GATTCAGTCGTCCAGG Endocrine
#> GCAAACTCAGACAAAT GCAAACTCAGACAAAT Endocrine
#> GCAATCACAGCTGCTG GCAATCACAGCTGCTG Endocrine
#> GCAATCATCCAACCAA GCAATCATCCAACCAA Ductal
#> GCACTCTCAGCGTCCA GCACTCTCAGCGTCCA Endocrine
#> GCAGCCACAGCGTTCG GCAGCCACAGCGTTCG Ngn3-high-EP
#> GCAGTTACATTTCACT GCAGTTACATTTCACT Pre-endocrine
#> GCAGTTATCTTGAGAC GCAGTTATCTTGAGAC Ductal
#> GCATGATAGCCTTGAT GCATGATAGCCTTGAT Ngn3-high-EP
#> GCATGATCAAGCGAGT GCATGATCAAGCGAGT Endocrine
#> GCATGATGTCACTGGC GCATGATGTCACTGGC Endocrine
#> GCATGATGTTTAGGAA GCATGATGTTTAGGAA Endocrine
#> GCATGATTCAAACGGG GCATGATTCAAACGGG Ductal
#> GCATGCGAGGAACTGC GCATGCGAGGAACTGC Pre-endocrine
#> GCATGCGCAGCTCCGA GCATGCGCAGCTCCGA Endocrine
#> GCATGTAAGAATCTCC GCATGTAAGAATCTCC Endocrine
#> GCATGTAAGCGTGTCC GCATGTAAGCGTGTCC Endocrine
#> GCATGTAGTTGCGCAC GCATGTAGTTGCGCAC Endocrine
#> GCATGTATCGTGGACC GCATGTATCGTGGACC Endocrine
#> GCCTCTAAGCTAGTCT GCCTCTAAGCTAGTCT Endocrine
#> GCCTCTAAGTCCGGTC GCCTCTAAGTCCGGTC Ngn3-high-EP
#> GCCTCTATCGCTTAGA GCCTCTATCGCTTAGA Ngn3-low-EP
#> GCGACCATCACCAGGC GCGACCATCACCAGGC Endocrine
#> GCGAGAAGTGGGTATG GCGAGAAGTGGGTATG Endocrine
#> GCGCAACGTTTCGCTC GCGCAACGTTTCGCTC Endocrine
#> GCGCAACTCACAACGT GCGCAACTCACAACGT Ductal
#> GCGCAGTGTGCTAGCC GCGCAGTGTGCTAGCC Pre-endocrine
#> GCGCCAAAGCTCCTTC GCGCCAAAGCTCCTTC Endocrine
#> GCGCCAATCTTATCTG GCGCCAATCTTATCTG Pre-endocrine
#> GCGCGATGTCCGAATT GCGCGATGTCCGAATT Endocrine
#> GCGGGTTAGACTTGAA GCGGGTTAGACTTGAA Ngn3-high-EP
#> GCGGGTTCAAGTAATG GCGGGTTCAAGTAATG Endocrine
#> GCGGGTTGTTACTGAC GCGGGTTGTTACTGAC Ductal
#> GCTCCTAAGGGCATGT GCTCCTAAGGGCATGT Endocrine
#> GCTCTGTGTCCATGAT GCTCTGTGTCCATGAT Pre-endocrine
#> GCTGCAGGTTACCGAT GCTGCAGGTTACCGAT Ductal
#> GCTGCGAAGAACAACT GCTGCGAAGAACAACT Endocrine
#> GCTGCGAAGCTAGCCC GCTGCGAAGCTAGCCC Ngn3-low-EP
#> GCTGCGAAGTATTGGA GCTGCGAAGTATTGGA Ngn3-high-EP
#> GCTGCGATCCCGGATG GCTGCGATCCCGGATG Pre-endocrine
#> GCTGCGATCGGCTACG GCTGCGATCGGCTACG Endocrine
#> GCTGCTTGTCTTCAAG GCTGCTTGTCTTCAAG Ductal
#> GCTGGGTGTCATACTG GCTGGGTGTCATACTG Endocrine
#> GCTGGGTGTCTAGAGG GCTGGGTGTCTAGAGG Ngn3-high-EP
#> GCTTCCAAGACATAAC GCTTCCAAGACATAAC Endocrine
#> GCTTCCAAGTTAAGTG GCTTCCAAGTTAAGTG Endocrine
#> GCTTGAAGTCAACTGT GCTTGAAGTCAACTGT Endocrine
#> GGAAAGCCAATGTAAG GGAAAGCCAATGTAAG Pre-endocrine
#> GGAAAGCGTCTTGATG GGAAAGCGTCTTGATG Ductal
#> GGAATAAGTAAGAGAG GGAATAAGTAAGAGAG Endocrine
#> GGAATAAGTTCGGGCT GGAATAAGTTCGGGCT Ductal
#> GGACAAGGTGATAAGT GGACAAGGTGATAAGT Ductal
#> GGACAAGTCAAAGACA GGACAAGTCAAAGACA Ngn3-high-EP
#> GGACAGAAGGCCGAAT GGACAGAAGGCCGAAT Endocrine
#> GGACAGACAATCCAAC GGACAGACAATCCAAC Pre-endocrine
#> GGACAGATCAGGCGAA GGACAGATCAGGCGAA Endocrine
#> GGACAGATCCTGCAGG GGACAGATCCTGCAGG Ductal
#> GGACAGATCCTTTCGG GGACAGATCCTTTCGG Ngn3-high-EP
#> GGACATTGTCTCCCTA GGACATTGTCTCCCTA Endocrine
#> GGACGTCAGGGTGTTG GGACGTCAGGGTGTTG Pre-endocrine
#> GGACGTCCACGCATCG GGACGTCCACGCATCG Ngn3-low-EP
#> GGACGTCCATGGATGG GGACGTCCATGGATGG Ngn3-high-EP
#> GGACGTCGTCAGAAGC GGACGTCGTCAGAAGC Pre-endocrine
#> GGACGTCGTCGAGATG GGACGTCGTCGAGATG Endocrine
#> GGACGTCGTCTAGGTT GGACGTCGTCTAGGTT Endocrine
#> GGACGTCGTTTAGCTG GGACGTCGTTTAGCTG Ductal
#> GGAGCAAAGCTTCGCG GGAGCAAAGCTTCGCG Ductal
#> GGAGCAAGTATCACCA GGAGCAAGTATCACCA Pre-endocrine
#> GGATGTTAGTAAGTAC GGATGTTAGTAAGTAC Endocrine
#> GGATGTTTCCAGTATG GGATGTTTCCAGTATG Ngn3-high-EP
#> GGCAATTCACGGCTAC GGCAATTCACGGCTAC Endocrine
#> GGCAATTCATCTCCCA GGCAATTCATCTCCCA Ngn3-high-EP
#> GGCAATTGTAGAGCTG GGCAATTGTAGAGCTG Endocrine
#> GGCCGATTCACTCTTA GGCCGATTCACTCTTA Ngn3-low-EP
#> GGCGACTGTAAGTTCC GGCGACTGTAAGTTCC Endocrine
#> GGCGTGTCACCAGGTC GGCGTGTCACCAGGTC Ngn3-high-EP
#> GGCGTGTCACCGATAT GGCGTGTCACCGATAT Pre-endocrine
#> GGCGTGTTCAGCTTAG GGCGTGTTCAGCTTAG Endocrine
#> GGCGTGTTCGAGAGCA GGCGTGTTCGAGAGCA Pre-endocrine
#> GGCTCGAAGATCACGG GGCTCGAAGATCACGG Pre-endocrine
#> GGCTCGAAGTGACTCT GGCTCGAAGTGACTCT Endocrine
#> GGCTCGACAGCTTCGG GGCTCGACAGCTTCGG Endocrine
#> GGCTGGTAGTCACGCC GGCTGGTAGTCACGCC Ngn3-high-EP
#> GGCTGGTTCATGCATG GGCTGGTTCATGCATG Ngn3-high-EP
#> GGGAATGAGCACACAG GGGAATGAGCACACAG Endocrine
#> GGGAATGAGTGTACGG GGGAATGAGTGTACGG Ductal
#> GGGAATGCAAAGTGCG GGGAATGCAAAGTGCG Endocrine
#> GGGAATGTCACAAACC GGGAATGTCACAAACC Ngn3-high-EP
#> GGGACCTCACTGTCGG GGGACCTCACTGTCGG Pre-endocrine
#> GGGACCTTCGAATGCT GGGACCTTCGAATGCT Ductal
#> GGGAGATAGATGTGGC GGGAGATAGATGTGGC Endocrine
#> GGGAGATAGCGAAGGG GGGAGATAGCGAAGGG Endocrine
#> GGGAGATAGTGACATA GGGAGATAGTGACATA Endocrine
#> GGGATGAAGGGCATGT GGGATGAAGGGCATGT Endocrine
#> GGGATGAGTCTGCGGT GGGATGAGTCTGCGGT Endocrine
#> GGGATGAGTTGTGGAG GGGATGAGTTGTGGAG Ngn3-high-EP
#> GGGCACTCATTTCAGG GGGCACTCATTTCAGG Ductal
#> GGGCACTGTCTCCATC GGGCACTGTCTCCATC Pre-endocrine
#> GGGCACTGTGGCAAAC GGGCACTGTGGCAAAC Ductal
#> GGGCATCCATGACATC GGGCATCCATGACATC Ductal
#> GGGCATCCATTTCAGG GGGCATCCATTTCAGG Endocrine
#> GGGCATCGTGGTCTCG GGGCATCGTGGTCTCG Ngn3-high-EP
#> GGGTCTGCACAGGCCT GGGTCTGCACAGGCCT Ngn3-high-EP
#> GGGTCTGCATGAAGTA GGGTCTGCATGAAGTA Ngn3-high-EP
#> GGGTCTGTCATGTGGT GGGTCTGTCATGTGGT Pre-endocrine
#> GGGTCTGTCCATGAAC GGGTCTGTCCATGAAC Endocrine
#> GGGTTGCCAAGCTGAG GGGTTGCCAAGCTGAG Ngn3-low-EP
#> GGGTTGCTCGAACGGA GGGTTGCTCGAACGGA Ductal
#> GGTATTGGTACTCGCG GGTATTGGTACTCGCG Ngn3-high-EP
#> GGTATTGGTCCCGACA GGTATTGGTCCCGACA Ductal
#> GGTGTTAAGGATCGCA GGTGTTAAGGATCGCA Ductal
#> GGTGTTAGTTCCTCCA GGTGTTAGTTCCTCCA Ductal
#> GTAACGTGTCACTGGC GTAACGTGTCACTGGC Ductal
#> GTAACTGGTCACTGGC GTAACTGGTCACTGGC Pre-endocrine
#> GTAACTGTCAGTGCAT GTAACTGTCAGTGCAT Ductal
#> GTACGTAGTCTCTTTA GTACGTAGTCTCTTTA Endocrine
#> GTACTCCCACAGGTTT GTACTCCCACAGGTTT Pre-endocrine
#> GTACTCCCACGTCTCT GTACTCCCACGTCTCT Ngn3-low-EP
#> GTACTCCTCCAACCAA GTACTCCTCCAACCAA Ngn3-high-EP
#> GTACTTTGTACCGTAT GTACTTTGTACCGTAT Endocrine
#> GTACTTTTCTTCGGTC GTACTTTTCTTCGGTC Endocrine
#> GTAGGCCAGCTAACAA GTAGGCCAGCTAACAA Pre-endocrine
#> GTAGGCCGTTACCAGT GTAGGCCGTTACCAGT Endocrine
#> GTAGTCACATCGATGT GTAGTCACATCGATGT Ngn3-high-EP
#> GTAGTCAGTCCGACGT GTAGTCAGTCCGACGT Ngn3-high-EP
#> GTATCTTCAGTGGGAT GTATCTTCAGTGGGAT Ductal
#> GTATCTTCATCGATGT GTATCTTCATCGATGT Ngn3-high-EP
#> GTATCTTGTACTCAAC GTATCTTGTACTCAAC Endocrine
#> GTATCTTGTCGCGGTT GTATCTTGTCGCGGTT Ngn3-high-EP
#> GTATCTTGTGGCCCTA GTATCTTGTGGCCCTA Ngn3-high-EP
#> GTATCTTTCGTGGACC GTATCTTTCGTGGACC Pre-endocrine
#> GTATTCTCACATTAGC GTATTCTCACATTAGC Ductal
#> GTATTCTCACTTGGAT GTATTCTCACTTGGAT Ductal
#> GTATTCTGTCCGTCAG GTATTCTGTCCGTCAG Ngn3-low-EP
#> GTATTCTTCTCTAAGG GTATTCTTCTCTAAGG Endocrine
#> GTCAAGTGTGGTCCGT GTCAAGTGTGGTCCGT Ductal
#> GTCACAACACGTCTCT GTCACAACACGTCTCT Ngn3-high-EP
#> GTCACGGAGCTATGCT GTCACGGAGCTATGCT Endocrine
#> GTCACGGCATCCGTGG GTCACGGCATCCGTGG Pre-endocrine
#> GTCACGGTCTTGCCGT GTCACGGTCTTGCCGT Pre-endocrine
#> GTCATTTGTCGTCTTC GTCATTTGTCGTCTTC Endocrine
#> GTCCTCACACCACGTG GTCCTCACACCACGTG Endocrine
#> GTCCTCATCATTTGGG GTCCTCATCATTTGGG Ngn3-high-EP
#> GTCCTCATCTGGAGCC GTCCTCATCTGGAGCC Ngn3-high-EP
#> GTCGGGTCAGGCTGAA GTCGGGTCAGGCTGAA Endocrine
#> GTCGTAAAGTTTAGGA GTCGTAAAGTTTAGGA Endocrine
#> GTCTCGTAGGCTAGAC GTCTCGTAGGCTAGAC Ngn3-high-EP
#> GTCTCGTCATAGTAAG GTCTCGTCATAGTAAG Ngn3-high-EP
#> GTCTCGTTCACTTCAT GTCTCGTTCACTTCAT Ngn3-high-EP
#> GTCTCGTTCTGCGTAA GTCTCGTTCTGCGTAA Endocrine
#> GTCTTCGTCCGTTGCT GTCTTCGTCCGTTGCT Endocrine
#> GTCTTCGTCTACCAGA GTCTTCGTCTACCAGA Pre-endocrine
#> GTGCATAAGAACAATC GTGCATAAGAACAATC Pre-endocrine
#> GTGCATAAGCTCCTCT GTGCATAAGCTCCTCT Ductal
#> GTGCATAGTTCAACCA GTGCATAGTTCAACCA Endocrine
#> GTGCATATCAAGGCTT GTGCATATCAAGGCTT Endocrine
#> GTGCGGTAGTAGGCCA GTGCGGTAGTAGGCCA Ductal
#> GTGCGGTCAGCGTAAG GTGCGGTCAGCGTAAG Ngn3-high-EP
#> GTGCTTCAGAGGGATA GTGCTTCAGAGGGATA Endocrine
#> GTGCTTCTCACGACTA GTGCTTCTCACGACTA Endocrine
#> GTGGGTCCATAAAGGT GTGGGTCCATAAAGGT Endocrine
#> GTGGGTCCATCGATTG GTGGGTCCATCGATTG Endocrine
#> GTGGGTCGTCCAGTAT GTGGGTCGTCCAGTAT Endocrine
#> GTGGGTCTCCGAATGT GTGGGTCTCCGAATGT Endocrine
#> GTGTGCGGTGTAAGTA GTGTGCGGTGTAAGTA Endocrine
#> GTGTTAGCACCATCCT GTGTTAGCACCATCCT Ductal
#> GTGTTAGGTGAACCTT GTGTTAGGTGAACCTT Pre-endocrine
#> GTTAAGCAGGATTCGG GTTAAGCAGGATTCGG Ngn3-high-EP
#> GTTACAGTCCGATATG GTTACAGTCCGATATG Ngn3-high-EP
#> GTTCATTAGAAGATTC GTTCATTAGAAGATTC Ngn3-low-EP
#> GTTCATTAGGCAGTCA GTTCATTAGGCAGTCA Pre-endocrine
#> GTTCATTTCCGAACGC GTTCATTTCCGAACGC Endocrine
#> GTTCTCGAGAATTCCC GTTCTCGAGAATTCCC Endocrine
#> GTTCTCGCAAGAAAGG GTTCTCGCAAGAAAGG Endocrine
#> GTTCTCGCAGACACTT GTTCTCGCAGACACTT Ductal
#> GTTTCTAAGAAGATTC GTTTCTAAGAAGATTC Endocrine
#> GTTTCTACAGGACCCT GTTTCTACAGGACCCT Endocrine
#> TAAGAGATCTGCGACG TAAGAGATCTGCGACG Pre-endocrine
#> TAAGTGCAGGGTTTCT TAAGTGCAGGGTTTCT Ductal
#> TAAGTGCTCCAAATGC TAAGTGCTCCAAATGC Endocrine
#> TACACGACAACGATCT TACACGACAACGATCT Ductal
#> TACACGACATGTAGTC TACACGACATGTAGTC Endocrine
#> TACACGAGTGCAACTT TACACGAGTGCAACTT Ngn3-high-EP
#> TACAGTGCAATCTACG TACAGTGCAATCTACG Ngn3-low-EP
#> TACAGTGCACAGGCCT TACAGTGCACAGGCCT Endocrine
#> TACAGTGGTAAACGCG TACAGTGGTAAACGCG Pre-endocrine
#> TACCTTAAGTACATGA TACCTTAAGTACATGA Ductal
#> TACCTTATCCGATATG TACCTTATCCGATATG Endocrine
#> TACGGATAGAGCTGGT TACGGATAGAGCTGGT Ductal
#> TACGGATAGGCACATG TACGGATAGGCACATG Endocrine
#> TACGGATAGTTTAGGA TACGGATAGTTTAGGA Ngn3-low-EP
#> TACGGATGTGTTCGAT TACGGATGTGTTCGAT Ductal
#> TACGGATTCCAAGTAC TACGGATTCCAAGTAC Ngn3-high-EP
#> TACGGGCAGCCAACAG TACGGGCAGCCAACAG Endocrine
#> TACGGGCAGCGTCAAG TACGGGCAGCGTCAAG Ductal
#> TACGGGCTCTTCGGTC TACGGGCTCTTCGGTC Endocrine
#> TACGGTACATGTTCCC TACGGTACATGTTCCC Ductal
#> TACTCATAGGTGTTAA TACTCATAGGTGTTAA Pre-endocrine
#> TACTCATCAGATCCAT TACTCATCAGATCCAT Endocrine
#> TACTCGCAGTAAGTAC TACTCGCAGTAAGTAC Endocrine
#> TACTCGCCAGATCGGA TACTCGCCAGATCGGA Endocrine
#> TACTCGCGTCGGCATC TACTCGCGTCGGCATC Endocrine
#> TACTTACCAACTGCGC TACTTACCAACTGCGC Ductal
#> TACTTACCATGGGACA TACTTACCATGGGACA Ngn3-high-EP
#> TACTTGTAGACAAAGG TACTTGTAGACAAAGG Endocrine
#> TAGACCAAGGAATCGC TAGACCAAGGAATCGC Ductal
#> TAGAGCTGTATGAAAC TAGAGCTGTATGAAAC Ductal
#> TAGAGCTGTTGTCGCG TAGAGCTGTTGTCGCG Ductal
#> TAGCCGGTCAACACAC TAGCCGGTCAACACAC Ductal
#> TAGGCATTCCAAAGTC TAGGCATTCCAAAGTC Ngn3-low-EP
#> TAGTGGTCAACTGCGC TAGTGGTCAACTGCGC Ductal
#> TAGTGGTCATTCACTT TAGTGGTCATTCACTT Pre-endocrine
#> TAGTGGTTCCTCAACC TAGTGGTTCCTCAACC Pre-endocrine
#> TAGTTGGCACGTAAGG TAGTTGGCACGTAAGG Ngn3-high-EP
#> TAGTTGGCAGTAACGG TAGTTGGCAGTAACGG Pre-endocrine
#> TAGTTGGGTACCGTAT TAGTTGGGTACCGTAT Ductal
#> TAGTTGGTCTTGCAAG TAGTTGGTCTTGCAAG Endocrine
#> TATCAGGCACCAGATT TATCAGGCACCAGATT Ductal
#> TATCTCACAACTGCTA TATCTCACAACTGCTA Ductal
#> TATCTCACACGCGAAA TATCTCACACGCGAAA Endocrine
#> TATCTCACATTGTGCA TATCTCACATTGTGCA Endocrine
#> TATCTCAGTTCGCTAA TATCTCAGTTCGCTAA Ductal
#> TATGCCCAGAACAATC TATGCCCAGAACAATC Pre-endocrine
#> TATGCCCGTCTAGCCG TATGCCCGTCTAGCCG Ngn3-high-EP
#> TATGCCCTCTTACCTA TATGCCCTCTTACCTA Ngn3-low-EP
#> TATTACCTCGTTGCCT TATTACCTCGTTGCCT Endocrine
#> TCAACGATCAACCATG TCAACGATCAACCATG Pre-endocrine
#> TCAATCTCATCCGCGA TCAATCTCATCCGCGA Ngn3-high-EP
#> TCAATCTCATCTCCCA TCAATCTCATCTCCCA Ngn3-low-EP
#> TCAATCTGTCAAAGAT TCAATCTGTCAAAGAT Ngn3-low-EP
#> TCAATCTTCGAACGGA TCAATCTTCGAACGGA Endocrine
#> TCACAAGAGTGACATA TCACAAGAGTGACATA Endocrine
#> TCACAAGCAACGATGG TCACAAGCAACGATGG Endocrine
#> TCACAAGCAGACTCGC TCACAAGCAGACTCGC Endocrine
#> TCACGAAGTCCGAGTC TCACGAAGTCCGAGTC Ngn3-high-EP
#> TCACGAATCCTATGTT TCACGAATCCTATGTT Ngn3-low-EP
#> TCAGATGCACTCGACG TCAGATGCACTCGACG Pre-endocrine
#> TCAGATGGTACACCGC TCAGATGGTACACCGC Endocrine
#> TCAGCAACACCAGGCT TCAGCAACACCAGGCT Ductal
#> TCAGCAATCGCCGTGA TCAGCAATCGCCGTGA Pre-endocrine
#> TCAGCTCAGTCGTACT TCAGCTCAGTCGTACT Ngn3-low-EP
#> TCAGGTAAGGCGTACA TCAGGTAAGGCGTACA Ngn3-high-EP
#> TCAGGTAAGTCAATAG TCAGGTAAGTCAATAG Ductal
#> TCAGGTACAGTCACTA TCAGGTACAGTCACTA Endocrine
#> TCAGGTATCTGTCCGT TCAGGTATCTGTCCGT Endocrine
#> TCATTACGTCAGAAGC TCATTACGTCAGAAGC Ductal
#> TCATTTGCATTGGCGC TCATTTGCATTGGCGC Pre-endocrine
#> TCCACACCAGGCGATA TCCACACCAGGCGATA Endocrine
#> TCCCGATAGTCGTTTG TCCCGATAGTCGTTTG Pre-endocrine
#> TCCCGATCAACGATGG TCCCGATCAACGATGG Ngn3-high-EP
#> TCGAGGCAGAACAATC TCGAGGCAGAACAATC Endocrine
#> TCGAGGCAGGCATGGT TCGAGGCAGGCATGGT Pre-endocrine
#> TCGCGAGAGTACGCGA TCGCGAGAGTACGCGA Pre-endocrine
#> TCGCGAGAGTCCGTAT TCGCGAGAGTCCGTAT Endocrine
#> TCGCGAGCACTTCTGC TCGCGAGCACTTCTGC Pre-endocrine
#> TCGGGACAGAGAACAG TCGGGACAGAGAACAG Ngn3-high-EP
#> TCGGGACGTCTGCGGT TCGGGACGTCTGCGGT Endocrine
#> TCGTACCCACGCATCG TCGTACCCACGCATCG Ngn3-low-EP
#> TCGTACCCATATACGC TCGTACCCATATACGC Ductal
#> TCTATTGCACAGGAGT TCTATTGCACAGGAGT Ductal
#> TCTATTGTCCTATTCA TCTATTGTCCTATTCA Endocrine
#> TCTATTGTCGTATCAG TCTATTGTCGTATCAG Pre-endocrine
#> TCTCATAAGAATGTGT TCTCATAAGAATGTGT Pre-endocrine
#> TCTCATACAAATTGCC TCTCATACAAATTGCC Endocrine
#> TCTCATAGTGGACGAT TCTCATAGTGGACGAT Ngn3-high-EP
#> TCTCTAAAGCTGGAAC TCTCTAAAGCTGGAAC Endocrine
#> TCTCTAAGTCGCATCG TCTCTAAGTCGCATCG Ductal
#> TCTCTAAGTGGCAAAC TCTCTAAGTGGCAAAC Ngn3-high-EP
#> TCTCTAAGTTACCGAT TCTCTAAGTTACCGAT Ductal
#> TCTCTAATCCCAACGG TCTCTAATCCCAACGG Pre-endocrine
#> TCTGAGAGTCAAACTC TCTGAGAGTCAAACTC Ductal
#> TCTGGAACACACGCTG TCTGGAACACACGCTG Ductal
#> TCTGGAACACCTATCC TCTGGAACACCTATCC Endocrine
#> TCTGGAAGTCATCGGC TCTGGAAGTCATCGGC Endocrine
#> TCTGGAATCGGTGTCG TCTGGAATCGGTGTCG Ductal
#> TCTTTCCGTGCAACTT TCTTTCCGTGCAACTT Endocrine
#> TCTTTCCTCGTCTGCT TCTTTCCTCGTCTGCT Ngn3-low-EP
#> TGAAAGAGTTATCACG TGAAAGAGTTATCACG Ngn3-low-EP
#> TGACGGCAGGAATGGA TGACGGCAGGAATGGA Ductal
#> TGACGGCCATATGAGA TGACGGCCATATGAGA Endocrine
#> TGACTAGGTGTATGGG TGACTAGGTGTATGGG Ngn3-high-EP
#> TGACTAGTCCACGTGG TGACTAGTCCACGTGG Ngn3-low-EP
#> TGACTAGTCCTATTCA TGACTAGTCCTATTCA Ngn3-high-EP
#> TGACTTTAGCCAGAAC TGACTTTAGCCAGAAC Endocrine
#> TGACTTTAGCGTTGCC TGACTTTAGCGTTGCC Ductal
#> TGACTTTAGCGTTTAC TGACTTTAGCGTTTAC Ductal
#> TGAGAGGAGATCCCAT TGAGAGGAGATCCCAT Endocrine
#> TGAGAGGAGCCAGGAT TGAGAGGAGCCAGGAT Endocrine
#> TGAGAGGAGCCGTCGT TGAGAGGAGCCGTCGT Ngn3-high-EP
#> TGAGAGGAGGCGATAC TGAGAGGAGGCGATAC Endocrine
#> TGAGAGGAGGTAAACT TGAGAGGAGGTAAACT Endocrine
#> TGAGAGGCAGGAACGT TGAGAGGCAGGAACGT Endocrine
#> TGAGAGGGTAAGGGCT TGAGAGGGTAAGGGCT Ductal
#> TGAGCATGTTAAAGAC TGAGCATGTTAAAGAC Ngn3-high-EP
#> TGAGCATTCACCCGAG TGAGCATTCACCCGAG Ductal
#> TGAGCATTCGGGAGTA TGAGCATTCGGGAGTA Ductal
#> TGAGCCGTCATGTCCC TGAGCCGTCATGTCCC Ductal
#> TGAGCCGTCCGAACGC TGAGCCGTCCGAACGC Ductal
#> TGAGCCGTCCGCTGTT TGAGCCGTCCGCTGTT Ngn3-high-EP
#> TGAGGGAAGAGGTACC TGAGGGAAGAGGTACC Ngn3-high-EP
#> TGAGGGACAGACTCGC TGAGGGACAGACTCGC Ductal
#> TGATTTCAGTACGCCC TGATTTCAGTACGCCC Ngn3-high-EP
#> TGATTTCAGTGACTCT TGATTTCAGTGACTCT Ductal
#> TGATTTCGTGAGGGTT TGATTTCGTGAGGGTT Endocrine
#> TGCCAAACATGACATC TGCCAAACATGACATC Ngn3-high-EP
#> TGCCCATCACTGTGTA TGCCCATCACTGTGTA Ngn3-low-EP
#> TGCCCTAAGTCCGGTC TGCCCTAAGTCCGGTC Pre-endocrine
#> TGCGCAGGTACGCACC TGCGCAGGTACGCACC Ductal
#> TGCGCAGTCTGCCAGG TGCGCAGTCTGCCAGG Ngn3-low-EP
#> TGCGGGTAGACGCACA TGCGGGTAGACGCACA Ngn3-high-EP
#> TGCGGGTAGGGTTCCC TGCGGGTAGGGTTCCC Endocrine
#> TGCGGGTTCCACTCCA TGCGGGTTCCACTCCA Endocrine
#> TGCGGGTTCTCAAACG TGCGGGTTCTCAAACG Ductal
#> TGCGTGGAGGTAAACT TGCGTGGAGGTAAACT Ngn3-low-EP
#> TGCGTGGAGGTTCCTA TGCGTGGAGGTTCCTA Pre-endocrine
#> TGCGTGGAGTACACCT TGCGTGGAGTACACCT Endocrine
#> TGCGTGGGTCCCTTGT TGCGTGGGTCCCTTGT Pre-endocrine
#> TGCGTGGGTTACTGAC TGCGTGGGTTACTGAC Pre-endocrine
#> TGCTACCCAGGGCATA TGCTACCCAGGGCATA Ngn3-low-EP
#> TGCTACCCATAAGACA TGCTACCCATAAGACA Ngn3-high-EP
#> TGCTGCTAGGATGCGT TGCTGCTAGGATGCGT Ngn3-high-EP
#> TGCTGCTGTCCGTCAG TGCTGCTGTCCGTCAG Ductal
#> TGGACGCAGTGTTGAA TGGACGCAGTGTTGAA Ngn3-low-EP
#> TGGACGCTCAGGTAAA TGGACGCTCAGGTAAA Ductal
#> TGGACGCTCTTGTTTG TGGACGCTCTTGTTTG Pre-endocrine
#> TGGCCAGAGCTACCTA TGGCCAGAGCTACCTA Ductal
#> TGGCCAGCAAGGACTG TGGCCAGCAAGGACTG Endocrine
#> TGGCCAGCAATGAAAC TGGCCAGCAATGAAAC Pre-endocrine
#> TGGCCAGCACGAAAGC TGGCCAGCACGAAAGC Ductal
#> TGGCCAGGTCGGCATC TGGCCAGGTCGGCATC Ductal
#> TGGCGCATCACAACGT TGGCGCATCACAACGT Ductal
#> TGGCTGGGTACCCAAT TGGCTGGGTACCCAAT Endocrine
#> TGGGAAGCACGTAAGG TGGGAAGCACGTAAGG Pre-endocrine
#> TGGGAAGCATGACATC TGGGAAGCATGACATC Ductal
#> TGGGCGTGTGTGCGTC TGGGCGTGTGTGCGTC Endocrine
#> TGGTTAGCAGTCGTGC TGGTTAGCAGTCGTGC Pre-endocrine
#> TGGTTCCCACCGCTAG TGGTTCCCACCGCTAG Ductal
#> TGGTTCCGTCACCTAA TGGTTCCGTCACCTAA Ductal
#> TGTATTCCAATGACCT TGTATTCCAATGACCT Ductal
#> TGTCCCAAGTTTAGGA TGTCCCAAGTTTAGGA Ngn3-low-EP
#> TGTCCCACATTATCTC TGTCCCACATTATCTC Endocrine
#> TGTCCCAGTCCGTGAC TGTCCCAGTCCGTGAC Endocrine
#> TGTCCCATCTCTGCTG TGTCCCATCTCTGCTG Endocrine
#> TGTGGTACAAGACGTG TGTGGTACAAGACGTG Ductal
#> TGTGGTACACATCCGG TGTGGTACACATCCGG Endocrine
#> TGTGGTAGTGCCTGCA TGTGGTAGTGCCTGCA Endocrine
#> TGTGGTATCCAGTAGT TGTGGTATCCAGTAGT Ngn3-high-EP
#> TGTGTTTAGGAATGGA TGTGTTTAGGAATGGA Endocrine
#> TGTGTTTGTAGATTAG TGTGTTTGTAGATTAG Endocrine
#> TGTGTTTGTATGAATG TGTGTTTGTATGAATG Pre-endocrine
#> TTAACTCCAAAGAATC TTAACTCCAAAGAATC Ductal
#> TTAACTCCAGGATTGG TTAACTCCAGGATTGG Endocrine
#> TTAACTCGTAATCGTC TTAACTCGTAATCGTC Pre-endocrine
#> TTAGGCAGTTTGTTGG TTAGGCAGTTTGTTGG Pre-endocrine
#> TTATGCTCACCCAGTG TTATGCTCACCCAGTG Endocrine
#> TTCCCAGAGTTACCCA TTCCCAGAGTTACCCA Ngn3-high-EP
#> TTCCCAGGTATTCTCT TTCCCAGGTATTCTCT Ngn3-low-EP
#> TTCCCAGTCATAGCAC TTCCCAGTCATAGCAC Pre-endocrine
#> TTCGAAGAGTACGCCC TTCGAAGAGTACGCCC Ngn3-high-EP
#> TTCGGTCAGCTAGCCC TTCGGTCAGCTAGCCC Ngn3-high-EP
#> TTCGGTCAGGGCATGT TTCGGTCAGGGCATGT Endocrine
#> TTCGGTCGTATAGTAG TTCGGTCGTATAGTAG Endocrine
#> TTCGGTCTCGACCAGC TTCGGTCTCGACCAGC Pre-endocrine
#> TTCTACAAGATCGATA TTCTACAAGATCGATA Ngn3-low-EP
#> TTCTACACATAAAGGT TTCTACACATAAAGGT Pre-endocrine
#> TTCTACAGTACTCTCC TTCTACAGTACTCTCC Ngn3-low-EP
#> TTCTACATCGCTTAGA TTCTACATCGCTTAGA Endocrine
#> TTCTACATCTGCAGTA TTCTACATCTGCAGTA Ductal
#> TTCTCAAAGCCGCCTA TTCTCAAAGCCGCCTA Ductal
#> TTCTCAACAATAGAGT TTCTCAACAATAGAGT Ngn3-high-EP
#> TTCTCCTAGTCCATAC TTCTCCTAGTCCATAC Endocrine
#> TTCTCCTCAATGGTCT TTCTCCTCAATGGTCT Ductal
#> TTCTTAGTCAGTGTTG TTCTTAGTCAGTGTTG Endocrine
#> TTGAACGAGAGCAATT TTGAACGAGAGCAATT Endocrine
#> TTGAACGAGGAGTTTA TTGAACGAGGAGTTTA Ngn3-high-EP
#> TTGAACGGTGAGGGAG TTGAACGGTGAGGGAG Endocrine
#> TTGAACGGTGATGCCC TTGAACGGTGATGCCC Ductal
#> TTGACTTAGACAAGCC TTGACTTAGACAAGCC Ductal
#> TTGACTTGTTCCATGA TTGACTTGTTCCATGA Ngn3-high-EP
#> TTGACTTTCACCGGGT TTGACTTTCACCGGGT Pre-endocrine
#> TTGACTTTCAGCAACT TTGACTTTCAGCAACT Ductal
#> TTGACTTTCCAGTAGT TTGACTTTCCAGTAGT Ductal
#> TTGCCGTCACCGAATT TTGCCGTCACCGAATT Endocrine
#> TTGCCGTTCTCTGTCG TTGCCGTTCTCTGTCG Ductal
#> TTGCGTCCACCGATAT TTGCGTCCACCGATAT Ngn3-high-EP
#> TTGGAACAGAAGATTC TTGGAACAGAAGATTC Endocrine
#> TTGGAACGTATATGAG TTGGAACGTATATGAG Endocrine
#> TTGGCAAAGAGTAAGG TTGGCAAAGAGTAAGG Endocrine
#> TTGTAGGTCAAAGTAG TTGTAGGTCAAAGTAG Ngn3-high-EP
#> TTTACTGCAGCATACT TTTACTGCAGCATACT Ngn3-high-EP
#> TTTACTGGTGCTAGCC TTTACTGGTGCTAGCC Ductal
#> TTTATGCAGGTTACCT TTTATGCAGGTTACCT Endocrine
#> TTTATGCGTCGAACAG TTTATGCGTCGAACAG Ductal
#> TTTATGCTCCATTCTA TTTATGCTCCATTCTA Ductal
#> TTTCCTCCAGGATCGA TTTCCTCCAGGATCGA Endocrine
#> TTTCCTCGTGGCGAAT TTTCCTCGTGGCGAAT Pre-endocrine
#> TTTGCGCAGACAGACC TTTGCGCAGACAGACC Ductal
#> TTTGGTTAGACAGGCT TTTGGTTAGACAGGCT Endocrine
#> TTTGGTTCACCAGATT TTTGGTTCACCAGATT Ductal
#> TTTGGTTCACGAAGCA TTTGGTTCACGAAGCA Ngn3-high-EP
#> TTTGGTTTCCTTTCGG TTTGGTTTCCTTTCGG Endocrine
#> TTTGTCAAGAATGTGT TTTGTCAAGAATGTGT Pre-endocrine
#> TTTGTCATCGAATGCT TTTGTCATCGAATGCT Endocrine
#> TTTGTCATCTGTTTGT TTTGTCATCTGTTTGT Endocrine
#>
#> $feature_metadata
#> features features_uique duplicated feature_split index
#> AUCell_A AUCell_A AUCell_A FALSE NA 1
#> GSVA_A GSVA_A GSVA_A FALSE NA 2
#> Sox9 Sox9 Sox9 FALSE NA 3
#> Anxa2 Anxa2 Anxa2 FALSE NA 4
#> Bicc1 Bicc1 Bicc1 FALSE NA 5
#>
#> $enrichment
#> NULL
#>
#>
#> $g_tree
#> gTree[GRID.gTree.11663]
#>
#> $matrix_list
#> $matrix_list$CellType
#> Ductal Ngn3-high-EP Endocrine Ngn3-low-EP Pre-endocrine
#> AUCell_A -0.6898028 0.6814256 -0.2740940 -1.0721911 1.3546623
#> GSVA_A 0.5904666 0.5803752 -1.7700722 0.2694525 0.3297779
#> Sox9 1.4288723 -0.6125486 -0.7946666 0.6788737 -0.7005309
#> Anxa2 1.1467252 -0.6170844 -0.7867363 1.0374551 -0.7803596
#> Bicc1 1.1109781 -0.6764747 -0.7375937 1.0784037 -0.7753133
#> attr(,"scaled:center")
#> AUCell_A GSVA_A Sox9 Anxa2 Bicc1
#> 0.01920529 0.10872510 1.96420268 2.04845477 1.94117464
#> attr(,"scaled:scale")
#> AUCell_A GSVA_A Sox9 Anxa2 Bicc1
#> 0.0007646939 0.0184930194 2.1633376549 2.5428690690 2.3797100097
#>
#>
#> $feature_split
#> NULL
#>
#> $cell_metadata
#> cells CellType
#> AAACCTGAGCCTTGAT AAACCTGAGCCTTGAT Ductal
#> AAACCTGGTAAGTGGC AAACCTGGTAAGTGGC Ngn3-high-EP
#> AAACGGGAGATATGGT AAACGGGAGATATGGT Ductal
#> AAACGGGCAAAGAATC AAACGGGCAAAGAATC Endocrine
#> AAACGGGGTACAGTTC AAACGGGGTACAGTTC Endocrine
#> AAACGGGTCAGCTCTC AAACGGGTCAGCTCTC Ngn3-low-EP
#> AAACGGGTCGCATGGC AAACGGGTCGCATGGC Endocrine
#> AAAGATGAGTCATGCT AAAGATGAGTCATGCT Endocrine
#> AAAGATGAGTCTTGCA AAAGATGAGTCTTGCA Endocrine
#> AAAGCAAAGCTACCTA AAAGCAAAGCTACCTA Pre-endocrine
#> AAAGCAACACATGGGA AAAGCAACACATGGGA Ductal
#> AAAGCAAGTGCAGTAG AAAGCAAGTGCAGTAG Endocrine
#> AAATGCCAGACTAAGT AAATGCCAGACTAAGT Endocrine
#> AAATGCCCAATGGAAT AAATGCCCAATGGAAT Endocrine
#> AAATGCCCACCAACCG AAATGCCCACCAACCG Ngn3-high-EP
#> AAATGCCCATGCTAGT AAATGCCCATGCTAGT Ngn3-high-EP
#> AAATGCCGTGATAAGT AAATGCCGTGATAAGT Endocrine
#> AACACGTGTCGACTGC AACACGTGTCGACTGC Pre-endocrine
#> AACCATGCACAGGAGT AACCATGCACAGGAGT Ngn3-high-EP
#> AACCATGGTTCGGCAC AACCATGGTTCGGCAC Ngn3-high-EP
#> AACCATGGTTGAACTC AACCATGGTTGAACTC Pre-endocrine
#> AACGTTGGTGCCTTGG AACGTTGGTGCCTTGG Ductal
#> AACTCAGGTGCCTGTG AACTCAGGTGCCTGTG Ngn3-high-EP
#> AACTCAGGTTGGAGGT AACTCAGGTTGGAGGT Ductal
#> AACTCAGTCCTCGCAT AACTCAGTCCTCGCAT Ngn3-high-EP
#> AACTCAGTCTGGTTCC AACTCAGTCTGGTTCC Ductal
#> AACTCCCCAGTTCATG AACTCCCCAGTTCATG Endocrine
#> AACTCCCTCGAGAGCA AACTCCCTCGAGAGCA Ductal
#> AACTCTTCATTTGCCC AACTCTTCATTTGCCC Endocrine
#> AACTGGTGTGCCTGTG AACTGGTGTGCCTGTG Ductal
#> AACTGGTTCTTACCGC AACTGGTTCTTACCGC Ductal
#> AACTTTCCACAGTCGC AACTTTCCACAGTCGC Ngn3-high-EP
#> AACTTTCCACTGCCAG AACTTTCCACTGCCAG Ductal
#> AACTTTCGTGACCAAG AACTTTCGTGACCAAG Ngn3-low-EP
#> AACTTTCTCCAGATCA AACTTTCTCCAGATCA Endocrine
#> AAGACCTAGACCTTTG AAGACCTAGACCTTTG Endocrine
#> AAGACCTAGTAGGCCA AAGACCTAGTAGGCCA Ngn3-high-EP
#> AAGACCTCAGGTTTCA AAGACCTCAGGTTTCA Ductal
#> AAGCCGCAGGCAGGTT AAGCCGCAGGCAGGTT Ductal
#> AAGCCGCCAATGGACG AAGCCGCCAATGGACG Ngn3-high-EP
#> AAGGAGCAGTACATGA AAGGAGCAGTACATGA Endocrine
#> AAGGCAGAGCTGAAAT AAGGCAGAGCTGAAAT Ductal
#> AAGGCAGTCCGAGCCA AAGGCAGTCCGAGCCA Ngn3-low-EP
#> AAGGTTCAGATAGTCA AAGGTTCAGATAGTCA Endocrine
#> AAGGTTCAGGAGCGAG AAGGTTCAGGAGCGAG Endocrine
#> AAGGTTCGTCAACTGT AAGGTTCGTCAACTGT Pre-endocrine
#> AAGTCTGGTTGGGACA AAGTCTGGTTGGGACA Endocrine
#> AAGTCTGTCCGAGCCA AAGTCTGTCCGAGCCA Ngn3-high-EP
#> AATCCAGAGGGTTCCC AATCCAGAGGGTTCCC Pre-endocrine
#> AATCCAGTCAGGTTCA AATCCAGTCAGGTTCA Pre-endocrine
#> AATCCAGTCGGTCCGA AATCCAGTCGGTCCGA Pre-endocrine
#> AATCGGTCATATGGTC AATCGGTCATATGGTC Ngn3-high-EP
#> AATCGGTTCCAAACTG AATCGGTTCCAAACTG Ngn3-high-EP
#> AATCGGTTCGTATCAG AATCGGTTCGTATCAG Endocrine
#> ACACCAAGTATAGGTA ACACCAAGTATAGGTA Endocrine
#> ACACCCTAGGACAGCT ACACCCTAGGACAGCT Ngn3-high-EP
#> ACACCCTTCCCAGGTG ACACCCTTCCCAGGTG Pre-endocrine
#> ACACCGGCATCACGTA ACACCGGCATCACGTA Ductal
#> ACACCGGCATGTCCTC ACACCGGCATGTCCTC Ductal
#> ACACCGGTCATGTCTT ACACCGGTCATGTCTT Endocrine
#> ACACTGAGTCCATGAT ACACTGAGTCCATGAT Pre-endocrine
#> ACAGCCGAGAGAACAG ACAGCCGAGAGAACAG Pre-endocrine
#> ACAGCCGAGAGGTACC ACAGCCGAGAGGTACC Endocrine
#> ACAGCCGAGTGGTCCC ACAGCCGAGTGGTCCC Ductal
#> ACAGCCGCATCGTCGG ACAGCCGCATCGTCGG Ngn3-high-EP
#> ACAGCTAAGTCGTTTG ACAGCTAAGTCGTTTG Endocrine
#> ACAGCTATCAAGGCTT ACAGCTATCAAGGCTT Endocrine
#> ACAGCTATCAGCATGT ACAGCTATCAGCATGT Ductal
#> ACAGCTATCTGGGCCA ACAGCTATCTGGGCCA Ductal
#> ACATACGAGTAGGCCA ACATACGAGTAGGCCA Ngn3-high-EP
#> ACATACGGTCTCTCTG ACATACGGTCTCTCTG Pre-endocrine
#> ACATCAGAGAGACTTA ACATCAGAGAGACTTA Pre-endocrine
#> ACATCAGTCCTTGGTC ACATCAGTCCTTGGTC Ngn3-high-EP
#> ACCAGTAAGTGATCGG ACCAGTAAGTGATCGG Endocrine
#> ACCAGTAGTTCGTTGA ACCAGTAGTTCGTTGA Endocrine
#> ACCAGTATCGTTTAGG ACCAGTATCGTTTAGG Ngn3-high-EP
#> ACCGTAAGTTATCACG ACCGTAAGTTATCACG Ductal
#> ACCTTTATCCTGCAGG ACCTTTATCCTGCAGG Endocrine
#> ACGAGCCCATGAGCGA ACGAGCCCATGAGCGA Endocrine
#> ACGAGGAAGTTACCCA ACGAGGAAGTTACCCA Ngn3-low-EP
#> ACGAGGATCATCACCC ACGAGGATCATCACCC Endocrine
#> ACGAGGATCGCCGTGA ACGAGGATCGCCGTGA Pre-endocrine
#> ACGATACAGATGAGAG ACGATACAGATGAGAG Endocrine
#> ACGATACTCGCGCCAA ACGATACTCGCGCCAA Endocrine
#> ACGATGTTCGCAGGCT ACGATGTTCGCAGGCT Pre-endocrine
#> ACGCAGCCATTCTCAT ACGCAGCCATTCTCAT Pre-endocrine
#> ACGCAGCGTAATCACC ACGCAGCGTAATCACC Ngn3-high-EP
#> ACGCCAGGTTACAGAA ACGCCAGGTTACAGAA Endocrine
#> ACGCCAGTCCCAAGTA ACGCCAGTCCCAAGTA Ngn3-high-EP
#> ACGCCGAAGGGTGTTG ACGCCGAAGGGTGTTG Endocrine
#> ACGCCGAGTTTGCATG ACGCCGAGTTTGCATG Endocrine
#> ACGGCCACACGTGAGA ACGGCCACACGTGAGA Ductal
#> ACGGGCTGTGCGATAG ACGGGCTGTGCGATAG Ductal
#> ACGTCAAAGGTGTTAA ACGTCAAAGGTGTTAA Endocrine
#> ACGTCAACACTGCCAG ACGTCAACACTGCCAG Ductal
#> ACGTCAACAGACAAAT ACGTCAACAGACAAAT Ductal
#> ACTATCTGTCAGGACA ACTATCTGTCAGGACA Endocrine
#> ACTATCTTCAAGATCC ACTATCTTCAAGATCC Pre-endocrine
#> ACTGAACAGCCTCGTG ACTGAACAGCCTCGTG Ductal
#> ACTGAGTAGTGACTCT ACTGAGTAGTGACTCT Ductal
#> ACTGATGCATCAGTAC ACTGATGCATCAGTAC Ngn3-high-EP
#> ACTGATGCATGGTCTA ACTGATGCATGGTCTA Endocrine
#> ACTGATGCATGTCTCC ACTGATGCATGTCTCC Pre-endocrine
#> ACTGATGGTGAGGGTT ACTGATGGTGAGGGTT Ductal
#> ACTGATGTCCCTAACC ACTGATGTCCCTAACC Ngn3-low-EP
#> ACTGCTCAGGTGTTAA ACTGCTCAGGTGTTAA Pre-endocrine
#> ACTGTCCCAGTTTACG ACTGTCCCAGTTTACG Pre-endocrine
#> ACTGTCCTCGCTAGCG ACTGTCCTCGCTAGCG Ngn3-high-EP
#> ACTTACTAGAGGGCTT ACTTACTAGAGGGCTT Ngn3-high-EP
#> ACTTACTAGCCGGTAA ACTTACTAGCCGGTAA Ngn3-low-EP
#> ACTTACTCAATCAGAA ACTTACTCAATCAGAA Ngn3-high-EP
#> ACTTACTCACATGTGT ACTTACTCACATGTGT Endocrine
#> ACTTGTTAGTGAAGTT ACTTGTTAGTGAAGTT Endocrine
#> ACTTGTTAGTTAAGTG ACTTGTTAGTTAAGTG Endocrine
#> ACTTGTTCATGGTCAT ACTTGTTCATGGTCAT Endocrine
#> ACTTGTTTCACCCGAG ACTTGTTTCACCCGAG Endocrine
#> ACTTTCAAGGATATAC ACTTTCAAGGATATAC Ductal
#> ACTTTCACAACTGCTA ACTTTCACAACTGCTA Ngn3-high-EP
#> AGAATAGCAATCACAC AGAATAGCAATCACAC Endocrine
#> AGAATAGCAGTACACT AGAATAGCAGTACACT Ductal
#> AGACGTTCAATGGTCT AGACGTTCAATGGTCT Endocrine
#> AGACGTTTCTTACCTA AGACGTTTCTTACCTA Ngn3-low-EP
#> AGAGCGACAGTAAGAT AGAGCGACAGTAAGAT Endocrine
#> AGAGCGATCTCTTGAT AGAGCGATCTCTTGAT Ductal
#> AGAGCTTAGCCGATTT AGAGCTTAGCCGATTT Ductal
#> AGAGCTTAGGTTACCT AGAGCTTAGGTTACCT Ngn3-high-EP
#> AGAGCTTGTTAAAGAC AGAGCTTGTTAAAGAC Ductal
#> AGAGTGGAGGCGATAC AGAGTGGAGGCGATAC Endocrine
#> AGAGTGGCACCTCGGA AGAGTGGCACCTCGGA Pre-endocrine
#> AGAGTGGGTAGCACGA AGAGTGGGTAGCACGA Ductal
#> AGAGTGGGTCACCTAA AGAGTGGGTCACCTAA Ngn3-low-EP
#> AGATCTGTCCGTCAAA AGATCTGTCCGTCAAA Pre-endocrine
#> AGATTGCAGCGTGTCC AGATTGCAGCGTGTCC Endocrine
#> AGATTGCAGCTGTCTA AGATTGCAGCTGTCTA Ductal
#> AGATTGCAGTTCCACA AGATTGCAGTTCCACA Ngn3-high-EP
#> AGATTGCCACCAGGCT AGATTGCCACCAGGCT Ductal
#> AGCAGCCGTCGCATCG AGCAGCCGTCGCATCG Ngn3-high-EP
#> AGCAGCCGTGTAACGG AGCAGCCGTGTAACGG Ngn3-high-EP
#> AGCAGCCTCTTTAGTC AGCAGCCTCTTTAGTC Ngn3-low-EP
#> AGCATACCACAGCCCA AGCATACCACAGCCCA Endocrine
#> AGCATACGTCCGCTGA AGCATACGTCCGCTGA Endocrine
#> AGCGGTCGTACCGAGA AGCGGTCGTACCGAGA Ngn3-high-EP
#> AGCGGTCGTCGAATCT AGCGGTCGTCGAATCT Ngn3-high-EP
#> AGCGGTCGTGTATGGG AGCGGTCGTGTATGGG Ductal
#> AGCGTCGCACAGGCCT AGCGTCGCACAGGCCT Ngn3-high-EP
#> AGCTCCTCAGTTTACG AGCTCCTCAGTTTACG Pre-endocrine
#> AGCTCCTGTGTGCCTG AGCTCCTGTGTGCCTG Ductal
#> AGCTCCTTCAATACCG AGCTCCTTCAATACCG Ngn3-high-EP
#> AGCTCCTTCGCCCTTA AGCTCCTTCGCCCTTA Pre-endocrine
#> AGCTCTCAGACATAAC AGCTCTCAGACATAAC Pre-endocrine
#> AGCTCTCGTTCAGTAC AGCTCTCGTTCAGTAC Endocrine
#> AGCTTGAAGACACGAC AGCTTGAAGACACGAC Ngn3-high-EP
#> AGCTTGAAGAGTGACC AGCTTGAAGAGTGACC Endocrine
#> AGCTTGACAATAGAGT AGCTTGACAATAGAGT Ngn3-low-EP
#> AGCTTGACAGTAAGAT AGCTTGACAGTAAGAT Ductal
#> AGCTTGACATCTATGG AGCTTGACATCTATGG Endocrine
#> AGCTTGAGTGCAGTAG AGCTTGAGTGCAGTAG Endocrine
#> AGGCCACAGCTGTCTA AGGCCACAGCTGTCTA Ngn3-low-EP
#> AGGCCACCATGACATC AGGCCACCATGACATC Endocrine
#> AGGCCGTCACGACGAA AGGCCGTCACGACGAA Ngn3-high-EP
#> AGGCCGTCACGGTGTC AGGCCGTCACGGTGTC Endocrine
#> AGGCCGTGTACCAGTT AGGCCGTGTACCAGTT Ngn3-low-EP
#> AGGGAGTAGGGTATCG AGGGAGTAGGGTATCG Ngn3-high-EP
#> AGGGAGTAGTGTCTCA AGGGAGTAGTGTCTCA Ductal
#> AGGGAGTTCCTCAATT AGGGAGTTCCTCAATT Endocrine
#> AGGGAGTTCGGAGGTA AGGGAGTTCGGAGGTA Endocrine
#> AGGGATGGTCGACTGC AGGGATGGTCGACTGC Endocrine
#> AGGGATGGTTAGGGTG AGGGATGGTTAGGGTG Ngn3-high-EP
#> AGGGATGTCGCATGAT AGGGATGTCGCATGAT Endocrine
#> AGGGATGTCGCATGGC AGGGATGTCGCATGGC Ngn3-high-EP
#> AGGGTGAGTGCGAAAC AGGGTGAGTGCGAAAC Endocrine
#> AGGTCATCACTTCTGC AGGTCATCACTTCTGC Endocrine
#> AGGTCATGTCTCCCTA AGGTCATGTCTCCCTA Endocrine
#> AGGTCATGTTTAGGAA AGGTCATGTTTAGGAA Pre-endocrine
#> AGTAGTCCACATGACT AGTAGTCCACATGACT Ngn3-low-EP
#> AGTAGTCGTGAGGGAG AGTAGTCGTGAGGGAG Endocrine
#> AGTAGTCGTTCTGTTT AGTAGTCGTTCTGTTT Ductal
#> AGTCTTTCACCAGGTC AGTCTTTCACCAGGTC Ductal
#> AGTCTTTCACGGACAA AGTCTTTCACGGACAA Endocrine
#> AGTCTTTGTTACGGAG AGTCTTTGTTACGGAG Endocrine
#> AGTGAGGGTTACGGAG AGTGAGGGTTACGGAG Ductal
#> AGTGAGGTCTCCAACC AGTGAGGTCTCCAACC Endocrine
#> AGTGGGACAGGTCGTC AGTGGGACAGGTCGTC Endocrine
#> AGTGGGAGTGGCAAAC AGTGGGAGTGGCAAAC Pre-endocrine
#> AGTGTCATCCCTTGTG AGTGTCATCCCTTGTG Pre-endocrine
#> AGTTGGTAGAGCCTAG AGTTGGTAGAGCCTAG Endocrine
#> AGTTGGTGTTGTCGCG AGTTGGTGTTGTCGCG Pre-endocrine
#> AGTTGGTTCACATAGC AGTTGGTTCACATAGC Ngn3-high-EP
#> ATAAGAGCATCGATTG ATAAGAGCATCGATTG Ductal
#> ATAGACCAGCGTGTCC ATAGACCAGCGTGTCC Endocrine
#> ATAGACCAGTGATCGG ATAGACCAGTGATCGG Endocrine
#> ATAGACCTCTAACTGG ATAGACCTCTAACTGG Ductal
#> ATCACGAGTGCACCAC ATCACGAGTGCACCAC Endocrine
#> ATCATCTGTGTTTGTG ATCATCTGTGTTTGTG Endocrine
#> ATCATCTTCTGTCAAG ATCATCTTCTGTCAAG Ductal
#> ATCATGGAGGAATGGA ATCATGGAGGAATGGA Endocrine
#> ATCATGGGTATTCGTG ATCATGGGTATTCGTG Endocrine
#> ATCATGGGTCCGCTGA ATCATGGGTCCGCTGA Ngn3-high-EP
#> ATCATGGTCCAGAGGA ATCATGGTCCAGAGGA Ductal
#> ATCCACCAGAGCCTAG ATCCACCAGAGCCTAG Endocrine
#> ATCCACCCAGCTCGCA ATCCACCCAGCTCGCA Endocrine
#> ATCCACCGTACCATCA ATCCACCGTACCATCA Ductal
#> ATCCGAAGTACACCGC ATCCGAAGTACACCGC Pre-endocrine
#> ATCCGAAGTGACTCAT ATCCGAAGTGACTCAT Ductal
#> ATCTGCCCAATCCGAT ATCTGCCCAATCCGAT Endocrine
#> ATCTGCCCATGTTGAC ATCTGCCCATGTTGAC Endocrine
#> ATGAGGGTCTTGACGA ATGAGGGTCTTGACGA Pre-endocrine
#> ATGTGTGCACAAGTAA ATGTGTGCACAAGTAA Ngn3-high-EP
#> ATGTGTGTCAAAGACA ATGTGTGTCAAAGACA Ngn3-high-EP
#> ATTACTCAGTTGCAGG ATTACTCAGTTGCAGG Ductal
#> ATTACTCTCAGGCGAA ATTACTCTCAGGCGAA Ductal
#> ATTACTCTCTGCCAGG ATTACTCTCTGCCAGG Endocrine
#> ATTATCCAGAAACCTA ATTATCCAGAAACCTA Ngn3-high-EP
#> ATTATCCAGACCCACC ATTATCCAGACCCACC Ngn3-high-EP
#> ATTATCCAGTGAACAT ATTATCCAGTGAACAT Ductal
#> ATTGGACAGTAATCCC ATTGGACAGTAATCCC Ngn3-low-EP
#> ATTGGTGTCCAGTATG ATTGGTGTCCAGTATG Ductal
#> ATTTCTGGTAGTGAAT ATTTCTGGTAGTGAAT Ngn3-low-EP
#> CAACCAAAGCGTTCCG CAACCAAAGCGTTCCG Pre-endocrine
#> CAACCAACAACCGCCA CAACCAACAACCGCCA Endocrine
#> CAACCAACACCTCGTT CAACCAACACCTCGTT Ngn3-high-EP
#> CAACCAATCGGGAGTA CAACCAATCGGGAGTA Ductal
#> CAAGAAAAGAGTACCG CAAGAAAAGAGTACCG Ngn3-high-EP
#> CAAGAAATCGTACCGG CAAGAAATCGTACCGG Ductal
#> CAAGGCCAGCTGTTCA CAAGGCCAGCTGTTCA Ductal
#> CAAGGCCAGTGTGGCA CAAGGCCAGTGTGGCA Ductal
#> CAAGGCCCACATTTCT CAAGGCCCACATTTCT Ngn3-high-EP
#> CAAGGCCTCAGTGCAT CAAGGCCTCAGTGCAT Endocrine
#> CAAGGCCTCTCAACTT CAAGGCCTCTCAACTT Pre-endocrine
#> CAAGGCCTCTTACCTA CAAGGCCTCTTACCTA Endocrine
#> CAAGTTGGTGTGCCTG CAAGTTGGTGTGCCTG Pre-endocrine
#> CAAGTTGTCTAACTGG CAAGTTGTCTAACTGG Pre-endocrine
#> CACAAACAGTAGCGGT CACAAACAGTAGCGGT Ngn3-high-EP
#> CACAAACCACGAAAGC CACAAACCACGAAAGC Endocrine
#> CACACAACAGCCAGAA CACACAACAGCCAGAA Ductal
#> CACACCTGTACATGTC CACACCTGTACATGTC Endocrine
#> CACACCTGTTATCCGA CACACCTGTTATCCGA Pre-endocrine
#> CACACTCAGATCCCAT CACACTCAGATCCCAT Ductal
#> CACACTCTCATGTGGT CACACTCTCATGTGGT Ngn3-low-EP
#> CACAGGCCAAGGCTCC CACAGGCCAAGGCTCC Pre-endocrine
#> CACAGGCCACGTCAGC CACAGGCCACGTCAGC Endocrine
#> CACAGTAGTGCACTTA CACAGTAGTGCACTTA Endocrine
#> CACAGTAGTTCCTCCA CACAGTAGTTCCTCCA Ductal
#> CACAGTATCCTCCTAG CACAGTATCCTCCTAG Ductal
#> CACATAGAGCGTGTCC CACATAGAGCGTGTCC Ductal
#> CACATTTGTTTCGCTC CACATTTGTTTCGCTC Ductal
#> CACATTTTCGAGGTAG CACATTTTCGAGGTAG Endocrine
#> CACATTTTCGCAGGCT CACATTTTCGCAGGCT Ductal
#> CACCACTAGCGATCCC CACCACTAGCGATCCC Endocrine
#> CACCACTGTGTGTGCC CACCACTGTGTGTGCC Pre-endocrine
#> CACCAGGCATAAAGGT CACCAGGCATAAAGGT Pre-endocrine
#> CACCTTGAGAGTAATC CACCTTGAGAGTAATC Ductal
#> CACTCCACATCGTCGG CACTCCACATCGTCGG Endocrine
#> CACTCCATCAACACAC CACTCCATCAACACAC Endocrine
#> CAGAATCCATTGGGCC CAGAATCCATTGGGCC Endocrine
#> CAGAATCGTCATTAGC CAGAATCGTCATTAGC Ngn3-high-EP
#> CAGAATCGTGTAAGTA CAGAATCGTGTAAGTA Ngn3-low-EP
#> CAGAGAGTCGGCCGAT CAGAGAGTCGGCCGAT Endocrine
#> CAGATCAAGCTGTTCA CAGATCAAGCTGTTCA Endocrine
#> CAGCAGCAGACAAAGG CAGCAGCAGACAAAGG Ductal
#> CAGCAGCCACACTGCG CAGCAGCCACACTGCG Ductal
#> CAGCAGCGTTCACGGC CAGCAGCGTTCACGGC Ductal
#> CAGCCGAAGCGATATA CAGCCGAAGCGATATA Ductal
#> CAGCCGAAGTAGGTGC CAGCCGAAGTAGGTGC Ductal
#> CAGCCGATCCTATTCA CAGCCGATCCTATTCA Ngn3-high-EP
#> CAGCCGATCGTTTATC CAGCCGATCGTTTATC Ngn3-high-EP
#> CAGCGACCACAGGAGT CAGCGACCACAGGAGT Ductal
#> CAGCTAAAGTCCATAC CAGCTAAAGTCCATAC Ductal
#> CAGCTAACATGCCTAA CAGCTAACATGCCTAA Pre-endocrine
#> CAGCTAAGTACCGAGA CAGCTAAGTACCGAGA Ngn3-high-EP
#> CAGCTGGCAAGTTGTC CAGCTGGCAAGTTGTC Pre-endocrine
#> CAGCTGGGTCTAACGT CAGCTGGGTCTAACGT Pre-endocrine
#> CAGCTGGGTTCTCATT CAGCTGGGTTCTCATT Endocrine
#> CAGGTGCAGCCAGGAT CAGGTGCAGCCAGGAT Endocrine
#> CAGGTGCCATCAGTCA CAGGTGCCATCAGTCA Pre-endocrine
#> CAGTCCTAGGTGGGTT CAGTCCTAGGTGGGTT Endocrine
#> CAGTCCTCAGGGCATA CAGTCCTCAGGGCATA Endocrine
#> CATATGGCATGTTGAC CATATGGCATGTTGAC Ductal
#> CATATGGGTTACTGAC CATATGGGTTACTGAC Endocrine
#> CATATGGTCACAGTAC CATATGGTCACAGTAC Pre-endocrine
#> CATATTCTCCTGTAGA CATATTCTCCTGTAGA Endocrine
#> CATCAGACATGCATGT CATCAGACATGCATGT Ductal
#> CATCAGAGTCTCTTTA CATCAGAGTCTCTTTA Pre-endocrine
#> CATCAGATCATCGCTC CATCAGATCATCGCTC Pre-endocrine
#> CATCGAAGTACGAAAT CATCGAAGTACGAAAT Endocrine
#> CATCGAAGTCACACGC CATCGAAGTCACACGC Endocrine
#> CATCGGGCACGTTGGC CATCGGGCACGTTGGC Endocrine
#> CATCGGGTCTTCGGTC CATCGGGTCTTCGGTC Ngn3-high-EP
#> CATGACACAACAACCT CATGACACAACAACCT Ngn3-low-EP
#> CATGACACATCGATTG CATGACACATCGATTG Ngn3-high-EP
#> CATGACAGTAGCTTGT CATGACAGTAGCTTGT Ngn3-low-EP
#> CATGACATCATGCATG CATGACATCATGCATG Endocrine
#> CATGCCTCACCCATTC CATGCCTCACCCATTC Endocrine
#> CATGGCGAGAGTCTGG CATGGCGAGAGTCTGG Ngn3-high-EP
#> CATTATCGTCATATCG CATTATCGTCATATCG Pre-endocrine
#> CCAATCCAGAGTAAGG CCAATCCAGAGTAAGG Ductal
#> CCACCTAAGACTTGAA CCACCTAAGACTTGAA Endocrine
#> CCACCTACAGCTCGAC CCACCTACAGCTCGAC Endocrine
#> CCACCTATCACCAGGC CCACCTATCACCAGGC Ductal
#> CCACCTATCTAAGCCA CCACCTATCTAAGCCA Endocrine
#> CCACGGAAGATCCTGT CCACGGAAGATCCTGT Endocrine
#> CCACGGAAGCAATCTC CCACGGAAGCAATCTC Endocrine
#> CCACGGAAGGGTGTTG CCACGGAAGGGTGTTG Endocrine
#> CCACGGAAGTCTCCTC CCACGGAAGTCTCCTC Pre-endocrine
#> CCACGGAGTCAACATC CCACGGAGTCAACATC Ngn3-high-EP
#> CCACGGAGTCTAGTCA CCACGGAGTCTAGTCA Endocrine
#> CCACGGAGTTACGACT CCACGGAGTTACGACT Ngn3-high-EP
#> CCACTACAGAGCTGGT CCACTACAGAGCTGGT Pre-endocrine
#> CCACTACAGAGTACCG CCACTACAGAGTACCG Ngn3-low-EP
#> CCACTACCAACGCACC CCACTACCAACGCACC Endocrine
#> CCAGCGACAGACGTAG CCAGCGACAGACGTAG Pre-endocrine
#> CCATGTCGTCGGCTCA CCATGTCGTCGGCTCA Ngn3-high-EP
#> CCATGTCTCCGCATAA CCATGTCTCCGCATAA Ngn3-high-EP
#> CCATTCGCATGCCTAA CCATTCGCATGCCTAA Ductal
#> CCATTCGGTAGGGACT CCATTCGGTAGGGACT Ductal
#> CCCAATCCAACTGGCC CCCAATCCAACTGGCC Endocrine
#> CCCAGTTAGCATCATC CCCAGTTAGCATCATC Endocrine
#> CCCAGTTGTCGTGGCT CCCAGTTGTCGTGGCT Endocrine
#> CCCAGTTGTGCGCTTG CCCAGTTGTGCGCTTG Endocrine
#> CCCAGTTTCAGCACAT CCCAGTTTCAGCACAT Endocrine
#> CCCATACAGACTTTCG CCCATACAGACTTTCG Endocrine
#> CCCATACCACATCCAA CCCATACCACATCCAA Ductal
#> CCCATACCATGCAACT CCCATACCATGCAACT Endocrine
#> CCCATACTCGTGGACC CCCATACTCGTGGACC Pre-endocrine
#> CCGGGATAGATCTGAA CCGGGATAGATCTGAA Ngn3-high-EP
#> CCGGGATTCGACAGCC CCGGGATTCGACAGCC Endocrine
#> CCGGTAGTCGGCTTGG CCGGTAGTCGGCTTGG Endocrine
#> CCGTACTAGGAGTACC CCGTACTAGGAGTACC Ngn3-high-EP
#> CCGTACTAGTCCGTAT CCGTACTAGTCCGTAT Ngn3-high-EP
#> CCGTACTTCAATAAGG CCGTACTTCAATAAGG Ngn3-low-EP
#> CCGTGGACATAGAAAC CCGTGGACATAGAAAC Endocrine
#> CCGTGGATCCAAGCCG CCGTGGATCCAAGCCG Ductal
#> CCGTGGATCTGTCTCG CCGTGGATCTGTCTCG Endocrine
#> CCGTGGATCTTAGAGC CCGTGGATCTTAGAGC Ngn3-high-EP
#> CCGTTCAAGCTAAACA CCGTTCAAGCTAAACA Endocrine
#> CCGTTCAAGGACCACA CCGTTCAAGGACCACA Ngn3-high-EP
#> CCGTTCACACATGACT CCGTTCACACATGACT Ngn3-low-EP
#> CCTACACAGATCCGAG CCTACACAGATCCGAG Ngn3-low-EP
#> CCTACACAGTACGTTC CCTACACAGTACGTTC Endocrine
#> CCTACACTCCTTTACA CCTACACTCCTTTACA Ductal
#> CCTACACTCTTTACAC CCTACACTCTTTACAC Endocrine
#> CCTACCACATCGATTG CCTACCACATCGATTG Ngn3-high-EP
#> CCTACCAGTTGCTCCT CCTACCAGTTGCTCCT Pre-endocrine
#> CCTACCATCTGCCCTA CCTACCATCTGCCCTA Endocrine
#> CCTAGCTCAGACTCGC CCTAGCTCAGACTCGC Ductal
#> CCTAGCTCATACTACG CCTAGCTCATACTACG Ductal
#> CCTAGCTGTGGCCCTA CCTAGCTGTGGCCCTA Endocrine
#> CCTAGCTTCGATGAGG CCTAGCTTCGATGAGG Ductal
#> CCTATTACAGACAAGC CCTATTACAGACAAGC Endocrine
#> CCTATTATCATCGCTC CCTATTATCATCGCTC Ngn3-high-EP
#> CCTCAGTAGTGCGATG CCTCAGTAGTGCGATG Pre-endocrine
#> CCTCAGTCAGACACTT CCTCAGTCAGACACTT Endocrine
#> CCTCAGTGTTACCAGT CCTCAGTGTTACCAGT Ngn3-high-EP
#> CCTCTGAGTATAAACG CCTCTGAGTATAAACG Ductal
#> CCTTACGAGATCACGG CCTTACGAGATCACGG Ductal
#> CCTTACGCACAACGCC CCTTACGCACAACGCC Pre-endocrine
#> CCTTCCCAGGCAGGTT CCTTCCCAGGCAGGTT Ductal
#> CCTTCCCCAAAGTGCG CCTTCCCCAAAGTGCG Ductal
#> CCTTCCCTCGTCGTTC CCTTCCCTCGTCGTTC Pre-endocrine
#> CCTTCGAAGATAGCAT CCTTCGAAGATAGCAT Endocrine
#> CCTTCGACAGCTGTTA CCTTCGACAGCTGTTA Ngn3-low-EP
#> CCTTCGATCACAGGCC CCTTCGATCACAGGCC Endocrine
#> CGAACATAGCCAGTAG CGAACATAGCCAGTAG Ductal
#> CGAACATCAGACGTAG CGAACATCAGACGTAG Ductal
#> CGAATGTAGTTCCACA CGAATGTAGTTCCACA Ductal
#> CGAATGTGTCTGGAGA CGAATGTGTCTGGAGA Pre-endocrine
#> CGAATGTTCACCGGGT CGAATGTTCACCGGGT Pre-endocrine
#> CGAATGTTCTTCATGT CGAATGTTCTTCATGT Endocrine
#> CGACCTTAGCGCTTAT CGACCTTAGCGCTTAT Ngn3-low-EP
#> CGACCTTAGGCTAGAC CGACCTTAGGCTAGAC Endocrine
#> CGACTTCCAGCCTATA CGACTTCCAGCCTATA Ductal
#> CGACTTCCAGCTGCAC CGACTTCCAGCTGCAC Pre-endocrine
#> CGACTTCTCACGCGGT CGACTTCTCACGCGGT Endocrine
#> CGAGAAGAGGGTTCCC CGAGAAGAGGGTTCCC Endocrine
#> CGAGAAGCACGCATCG CGAGAAGCACGCATCG Ngn3-high-EP
#> CGAGAAGTCGTTACAG CGAGAAGTCGTTACAG Endocrine
#> CGAGCACAGCTGAAAT CGAGCACAGCTGAAAT Ductal
#> CGAGCACAGGCAGGTT CGAGCACAGGCAGGTT Pre-endocrine
#> CGAGCACGTAGTAGTA CGAGCACGTAGTAGTA Pre-endocrine
#> CGAGCACTCTACCTGC CGAGCACTCTACCTGC Ductal
#> CGAGCACTCTTCGAGA CGAGCACTCTTCGAGA Ngn3-low-EP
#> CGATCGGAGCTGCAAG CGATCGGAGCTGCAAG Pre-endocrine
#> CGATCGGCACGCCAGT CGATCGGCACGCCAGT Ngn3-high-EP
#> CGATGGCGTTACGGAG CGATGGCGTTACGGAG Ductal
#> CGATGGCTCAGTGTTG CGATGGCTCAGTGTTG Endocrine
#> CGATGGCTCTGTGCAA CGATGGCTCTGTGCAA Ductal
#> CGATGTAAGTTTAGGA CGATGTAAGTTTAGGA Endocrine
#> CGATGTAGTACTCTCC CGATGTAGTACTCTCC Ngn3-high-EP
#> CGATTGAAGTCAAGGC CGATTGAAGTCAAGGC Pre-endocrine
#> CGCCAAGCAAAGTCAA CGCCAAGCAAAGTCAA Ngn3-high-EP
#> CGCCAAGCACCGGAAA CGCCAAGCACCGGAAA Ductal
#> CGCCAAGTCTAAGCCA CGCCAAGTCTAAGCCA Endocrine
#> CGCGGTAAGAGCTGGT CGCGGTAAGAGCTGGT Endocrine
#> CGCGGTAAGCTGCAAG CGCGGTAAGCTGCAAG Endocrine
#> CGCGTTTCAGTCTTCC CGCGTTTCAGTCTTCC Endocrine
#> CGCTGGAGTCCATCCT CGCTGGAGTCCATCCT Ductal
#> CGCTTCATCTCTGTCG CGCTTCATCTCTGTCG Endocrine
#> CGGACACAGTGTCTCA CGGACACAGTGTCTCA Ductal
#> CGGACGTAGACACGAC CGGACGTAGACACGAC Endocrine
#> CGGACTGAGGTAGCTG CGGACTGAGGTAGCTG Ductal
#> CGGACTGTCTTGAGAC CGGACTGTCTTGAGAC Endocrine
#> CGGAGCTAGATAGTCA CGGAGCTAGATAGTCA Endocrine
#> CGGAGCTCATTGGGCC CGGAGCTCATTGGGCC Ductal
#> CGGAGTCAGTTTAGGA CGGAGTCAGTTTAGGA Endocrine
#> CGGCTAGGTACTCGCG CGGCTAGGTACTCGCG Ngn3-high-EP
#> CGGGTCAAGGCGATAC CGGGTCAAGGCGATAC Endocrine
#> CGGGTCAAGGCTCAGA CGGGTCAAGGCTCAGA Ductal
#> CGGTTAAAGAGCTGCA CGGTTAAAGAGCTGCA Ductal
#> CGGTTAAGTAAACACA CGGTTAAGTAAACACA Ngn3-low-EP
#> CGGTTAAGTCTAGCCG CGGTTAAGTCTAGCCG Ngn3-high-EP
#> CGGTTAATCACCGGGT CGGTTAATCACCGGGT Pre-endocrine
#> CGGTTAATCGTCTGAA CGGTTAATCGTCTGAA Ductal
#> CGTAGCGTCGGAAATA CGTAGCGTCGGAAATA Pre-endocrine
#> CGTAGGCAGCGTAGTG CGTAGGCAGCGTAGTG Ductal
#> CGTCACTAGGGTGTGT CGTCACTAGGGTGTGT Ductal
#> CGTCACTCACGAAATA CGTCACTCACGAAATA Ngn3-low-EP
#> CGTCACTCATAGTAAG CGTCACTCATAGTAAG Ngn3-low-EP
#> CGTCACTTCACTTATC CGTCACTTCACTTATC Pre-endocrine
#> CGTCACTTCTAACTCT CGTCACTTCTAACTCT Endocrine
#> CGTCAGGAGCCTATGT CGTCAGGAGCCTATGT Ngn3-low-EP
#> CGTCAGGCATGGGAAC CGTCAGGCATGGGAAC Ngn3-low-EP
#> CGTCAGGGTAGGCATG CGTCAGGGTAGGCATG Ngn3-high-EP
#> CGTCAGGTCCGCAAGC CGTCAGGTCCGCAAGC Endocrine
#> CGTCAGGTCCTGCCAT CGTCAGGTCCTGCCAT Ductal
#> CGTCCATGTCTAACGT CGTCCATGTCTAACGT Endocrine
#> CGTCCATTCGCGCCAA CGTCCATTCGCGCCAA Ngn3-high-EP
#> CGTCTACAGATCTGCT CGTCTACAGATCTGCT Ductal
#> CGTCTACAGGGAGTAA CGTCTACAGGGAGTAA Endocrine
#> CGTGAGCTCAGCGATT CGTGAGCTCAGCGATT Ngn3-high-EP
#> CGTGAGCTCTTCTGGC CGTGAGCTCTTCTGGC Endocrine
#> CGTGTAAGTCTTGATG CGTGTAAGTCTTGATG Ngn3-low-EP
#> CGTGTAATCATTATCC CGTGTAATCATTATCC Ngn3-high-EP
#> CGTGTAATCCATGCTC CGTGTAATCCATGCTC Ductal
#> CGTGTCTCAGAAGCAC CGTGTCTCAGAAGCAC Endocrine
#> CGTGTCTGTAGCGTCC CGTGTCTGTAGCGTCC Endocrine
#> CGTTAGAAGTATCTCG CGTTAGAAGTATCTCG Ductal
#> CGTTAGAGTGCAACGA CGTTAGAGTGCAACGA Ductal
#> CGTTCTGAGGACGAAA CGTTCTGAGGACGAAA Endocrine
#> CGTTCTGTCACAATGC CGTTCTGTCACAATGC Ngn3-high-EP
#> CGTTCTGTCGATAGAA CGTTCTGTCGATAGAA Endocrine
#> CGTTGGGCAGGCGATA CGTTGGGCAGGCGATA Pre-endocrine
#> CTAACTTAGAGGTACC CTAACTTAGAGGTACC Endocrine
#> CTAACTTAGATCACGG CTAACTTAGATCACGG Endocrine
#> CTAACTTCATCTGGTA CTAACTTCATCTGGTA Endocrine
#> CTAACTTCATTGGGCC CTAACTTCATTGGGCC Ductal
#> CTAAGACAGCACGCCT CTAAGACAGCACGCCT Ngn3-high-EP
#> CTAAGACAGCCACGCT CTAAGACAGCCACGCT Ngn3-low-EP
#> CTAAGACGTATCAGTC CTAAGACGTATCAGTC Endocrine
#> CTAAGACTCTGCGGCA CTAAGACTCTGCGGCA Endocrine
#> CTAATGGCATGGTTGT CTAATGGCATGGTTGT Pre-endocrine
#> CTACACCCATCAGTCA CTACACCCATCAGTCA Endocrine
#> CTACACCGTCGAATCT CTACACCGTCGAATCT Ductal
#> CTACACCGTCGCCATG CTACACCGTCGCCATG Ductal
#> CTACATTCACCGTTGG CTACATTCACCGTTGG Pre-endocrine
#> CTACATTGTTACTGAC CTACATTGTTACTGAC Pre-endocrine
#> CTACCCAGTGCGCTTG CTACCCAGTGCGCTTG Ductal
#> CTACGTCAGCTAGGCA CTACGTCAGCTAGGCA Pre-endocrine
#> CTAGAGTCACTGTTAG CTAGAGTCACTGTTAG Ductal
#> CTAGCCTGTCTCTTAT CTAGCCTGTCTCTTAT Ductal
#> CTAGCCTGTTCGGGCT CTAGCCTGTTCGGGCT Ductal
#> CTAGCCTTCAGCGATT CTAGCCTTCAGCGATT Ductal
#> CTAGTGATCAAAGACA CTAGTGATCAAAGACA Endocrine
#> CTAGTGATCCCAGGTG CTAGTGATCCCAGGTG Endocrine
#> CTCACACGTCACACGC CTCACACGTCACACGC Ngn3-high-EP
#> CTCAGAACACTAGTAC CTCAGAACACTAGTAC Ngn3-high-EP
#> CTCAGAACATAGGATA CTCAGAACATAGGATA Endocrine
#> CTCAGAAGTCCTCTTG CTCAGAAGTCCTCTTG Ductal
#> CTCAGAATCAGGTTCA CTCAGAATCAGGTTCA Ngn3-low-EP
#> CTCATTACAAGCGCTC CTCATTACAAGCGCTC Ductal
#> CTCCTAGCACAACGTT CTCCTAGCACAACGTT Pre-endocrine
#> CTCGAAAGTAAGAGGA CTCGAAAGTAAGAGGA Ngn3-high-EP
#> CTCGAAAGTGGTGTAG CTCGAAAGTGGTGTAG Endocrine
#> CTCGAAATCCTGCCAT CTCGAAATCCTGCCAT Endocrine
#> CTCGAGGCAAGTCTGT CTCGAGGCAAGTCTGT Endocrine
#> CTCGAGGGTAAGTAGT CTCGAGGGTAAGTAGT Endocrine
#> CTCGAGGTCTGTTGAG CTCGAGGTCTGTTGAG Ngn3-high-EP
#> CTCGGAGGTCATTAGC CTCGGAGGTCATTAGC Pre-endocrine
#> CTCGGAGGTGAAGGCT CTCGGAGGTGAAGGCT Pre-endocrine
#> CTCGTACCAATGGATA CTCGTACCAATGGATA Ductal
#> CTCGTACTCTTTAGTC CTCGTACTCTTTAGTC Endocrine
#> CTCGTCAAGAACAATC CTCGTCAAGAACAATC Endocrine
#> CTCGTCACAACTGCTA CTCGTCACAACTGCTA Ductal
#> CTCGTCATCGGTCTAA CTCGTCATCGGTCTAA Ductal
#> CTCTAATAGTTATCGC CTCTAATAGTTATCGC Endocrine
#> CTCTGGTAGCAATCTC CTCTGGTAGCAATCTC Ngn3-high-EP
#> CTCTGGTAGTCCCACG CTCTGGTAGTCCCACG Ductal
#> CTCTGGTCAACACCTA CTCTGGTCAACACCTA Ductal
#> CTCTGGTCAAGACACG CTCTGGTCAAGACACG Endocrine
#> CTCTGGTCAAGTTGTC CTCTGGTCAAGTTGTC Pre-endocrine
#> CTCTGGTCAGTTCATG CTCTGGTCAGTTCATG Ngn3-low-EP
#> CTGAAACAGACTAGGC CTGAAACAGACTAGGC Ductal
#> CTGAAACGTAGCGCAA CTGAAACGTAGCGCAA Endocrine
#> CTGAAACGTATGGTTC CTGAAACGTATGGTTC Endocrine
#> CTGAAACTCGATAGAA CTGAAACTCGATAGAA Ngn3-high-EP
#> CTGAAACTCGCCGTGA CTGAAACTCGCCGTGA Ductal
#> CTGAAACTCTGTTTGT CTGAAACTCTGTTTGT Ngn3-high-EP
#> CTGAAGTGTTTGGGCC CTGAAGTGTTTGGGCC Endocrine
#> CTGAAGTTCTGCGGCA CTGAAGTTCTGCGGCA Pre-endocrine
#> CTGATCCTCAGTTGAC CTGATCCTCAGTTGAC Ductal
#> CTGCCTAGTCTCCATC CTGCCTAGTCTCCATC Ductal
#> CTGCGGACAGACTCGC CTGCGGACAGACTCGC Ngn3-high-EP
#> CTGCGGACATTAGCCA CTGCGGACATTAGCCA Pre-endocrine
#> CTGCGGATCAGCATGT CTGCGGATCAGCATGT Ductal
#> CTGCTGTAGACTGGGT CTGCTGTAGACTGGGT Pre-endocrine
#> CTGCTGTCAAAGCAAT CTGCTGTCAAAGCAAT Endocrine
#> CTGGTCTAGGATTCGG CTGGTCTAGGATTCGG Pre-endocrine
#> CTGGTCTAGGTGATTA CTGGTCTAGGTGATTA Endocrine
#> CTGGTCTCAGGAATGC CTGGTCTCAGGAATGC Ductal
#> CTGGTCTTCGCATGGC CTGGTCTTCGCATGGC Ngn3-high-EP
#> CTGTGCTCATTACCTT CTGTGCTCATTACCTT Ngn3-high-EP
#> CTTAACTCAAGCGTAG CTTAACTCAAGCGTAG Ductal
#> CTTAACTTCAGCTTAG CTTAACTTCAGCTTAG Pre-endocrine
#> CTTAGGACACAGGTTT CTTAGGACACAGGTTT Ductal
#> CTTAGGAGTCCGAATT CTTAGGAGTCCGAATT Ductal
#> CTTAGGATCAACGGGA CTTAGGATCAACGGGA Endocrine
#> CTTAGGATCACTCTTA CTTAGGATCACTCTTA Ngn3-high-EP
#> CTTCTCTAGGCAGGTT CTTCTCTAGGCAGGTT Ductal
#> CTTCTCTAGTCCGTAT CTTCTCTAGTCCGTAT Pre-endocrine
#> CTTCTCTCAGCATACT CTTCTCTCAGCATACT Endocrine
#> CTTCTCTGTCAGAGGT CTTCTCTGTCAGAGGT Endocrine
#> CTTGGCTAGACAGACC CTTGGCTAGACAGACC Ductal
#> CTTGGCTAGGACACCA CTTGGCTAGGACACCA Ductal
#> CTTGGCTCAAAGGAAG CTTGGCTCAAAGGAAG Pre-endocrine
#> CTTGGCTCAGCCACCA CTTGGCTCAGCCACCA Ngn3-high-EP
#> CTTGGCTCATCCCACT CTTGGCTCATCCCACT Endocrine
#> CTTGGCTGTTAGAACA CTTGGCTGTTAGAACA Endocrine
#> CTTTGCGAGACCACGA CTTTGCGAGACCACGA Pre-endocrine
#> CTTTGCGAGGAATGGA CTTTGCGAGGAATGGA Ngn3-low-EP
#> CTTTGCGTCCACGTGG CTTTGCGTCCACGTGG Endocrine
#> GAAACTCAGAAACCTA GAAACTCAGAAACCTA Endocrine
#> GAAATGAAGCTCCTCT GAAATGAAGCTCCTCT Ngn3-high-EP
#> GAAATGACAACTGGCC GAAATGACAACTGGCC Ngn3-low-EP
#> GAAATGACACATGACT GAAATGACACATGACT Ductal
#> GAAATGAGTCACCCAG GAAATGAGTCACCCAG Ngn3-high-EP
#> GAAATGAGTCGGATCC GAAATGAGTCGGATCC Ductal
#> GAACATCGTAATCGTC GAACATCGTAATCGTC Ductal
#> GAACCTAAGTAGATGT GAACCTAAGTAGATGT Endocrine
#> GAACCTATCACTTATC GAACCTATCACTTATC Endocrine
#> GAACGGAAGTCAAGCG GAACGGAAGTCAAGCG Endocrine
#> GAACGGACAAGGACAC GAACGGACAAGGACAC Endocrine
#> GAAGCAGGTCGCGAAA GAAGCAGGTCGCGAAA Pre-endocrine
#> GAAGCAGGTCTAGAGG GAAGCAGGTCTAGAGG Endocrine
#> GAAGCAGTCAAGGCTT GAAGCAGTCAAGGCTT Ductal
#> GAATGAAAGAATGTGT GAATGAAAGAATGTGT Ngn3-high-EP
#> GAATGAACAATCGAAA GAATGAACAATCGAAA Ductal
#> GAATGAACAGCCTTGG GAATGAACAGCCTTGG Ductal
#> GAATGAATCATCGGAT GAATGAATCATCGGAT Endocrine
#> GACACGCCAAAGGAAG GACACGCCAAAGGAAG Ductal
#> GACCAATAGAATGTGT GACCAATAGAATGTGT Endocrine
#> GACCAATCAGACGCCT GACCAATCAGACGCCT Pre-endocrine
#> GACCAATGTTATCCGA GACCAATGTTATCCGA Endocrine
#> GACCAATGTTGGACCC GACCAATGTTGGACCC Endocrine
#> GACCAATTCAACGGCC GACCAATTCAACGGCC Ngn3-high-EP
#> GACCTGGGTTCGTGAT GACCTGGGTTCGTGAT Endocrine
#> GACGCGTCAGTCGTGC GACGCGTCAGTCGTGC Ductal
#> GACGCGTGTCGGCTCA GACGCGTGTCGGCTCA Endocrine
#> GACGGCTAGGTGATAT GACGGCTAGGTGATAT Ductal
#> GACGGCTTCAACTCTT GACGGCTTCAACTCTT Pre-endocrine
#> GACGTGCGTCACCTAA GACGTGCGTCACCTAA Pre-endocrine
#> GACGTGCTCCGGCACA GACGTGCTCCGGCACA Endocrine
#> GACGTTAAGACGCAAC GACGTTAAGACGCAAC Ductal
#> GACGTTAAGACGCTTT GACGTTAAGACGCTTT Ngn3-high-EP
#> GACGTTACATCTCGCT GACGTTACATCTCGCT Endocrine
#> GACGTTAGTGTGAATA GACGTTAGTGTGAATA Pre-endocrine
#> GACTAACGTCATCCCT GACTAACGTCATCCCT Ngn3-high-EP
#> GACTAACTCAGTCCCT GACTAACTCAGTCCCT Ductal
#> GACTACAGTTCGTGAT GACTACAGTTCGTGAT Ductal
#> GACTGCGAGAATCTCC GACTGCGAGAATCTCC Ngn3-high-EP
#> GAGCAGAAGCGTGTCC GAGCAGAAGCGTGTCC Ductal
#> GAGCAGAGTTTGCATG GAGCAGAGTTTGCATG Endocrine
#> GAGCAGATCAACTCTT GAGCAGATCAACTCTT Pre-endocrine
#> GAGGTGAGTCCGCTGA GAGGTGAGTCCGCTGA Pre-endocrine
#> GAGGTGAGTGACAAAT GAGGTGAGTGACAAAT Ductal
#> GAGTCCGAGCGATGAC GAGTCCGAGCGATGAC Ngn3-high-EP
#> GAGTCCGAGTTAAGTG GAGTCCGAGTTAAGTG Ductal
#> GAGTCCGCAATTGCTG GAGTCCGCAATTGCTG Ductal
#> GATCAGTAGGTGATAT GATCAGTAGGTGATAT Endocrine
#> GATCGATAGAAAGTGG GATCGATAGAAAGTGG Pre-endocrine
#> GATCGATAGGATGGTC GATCGATAGGATGGTC Endocrine
#> GATCGATGTAGCGATG GATCGATGTAGCGATG Endocrine
#> GATCGATGTTGTTTGG GATCGATGTTGTTTGG Endocrine
#> GATCGCGCACCAGGCT GATCGCGCACCAGGCT Pre-endocrine
#> GATCGTAAGTAGGTGC GATCGTAAGTAGGTGC Pre-endocrine
#> GATCGTAGTCCTAGCG GATCGTAGTCCTAGCG Ductal
#> GATCTAGAGCTAGCCC GATCTAGAGCTAGCCC Endocrine
#> GATCTAGGTGCCTTGG GATCTAGGTGCCTTGG Ductal
#> GATGAAAAGTTGTAGA GATGAAAAGTTGTAGA Ngn3-low-EP
#> GATGAAACAGGCTGAA GATGAAACAGGCTGAA Endocrine
#> GATGAAACATGGTTGT GATGAAACATGGTTGT Ductal
#> GATGAGGAGTGGACGT GATGAGGAGTGGACGT Ngn3-high-EP
#> GATGAGGTCTGGGCCA GATGAGGTCTGGGCCA Ductal
#> GATGCTAGTAGAAGGA GATGCTAGTAGAAGGA Ductal
#> GATGCTAGTGTTAAGA GATGCTAGTGTTAAGA Pre-endocrine
#> GATTCAGAGCGATGAC GATTCAGAGCGATGAC Pre-endocrine
#> GATTCAGAGTACGTTC GATTCAGAGTACGTTC Endocrine
#> GATTCAGTCGCCAGCA GATTCAGTCGCCAGCA Pre-endocrine
#> GATTCAGTCGTCCAGG GATTCAGTCGTCCAGG Endocrine
#> GCAAACTCAGACAAAT GCAAACTCAGACAAAT Endocrine
#> GCAATCACAGCTGCTG GCAATCACAGCTGCTG Endocrine
#> GCAATCATCCAACCAA GCAATCATCCAACCAA Ductal
#> GCACTCTCAGCGTCCA GCACTCTCAGCGTCCA Endocrine
#> GCAGCCACAGCGTTCG GCAGCCACAGCGTTCG Ngn3-high-EP
#> GCAGTTACATTTCACT GCAGTTACATTTCACT Pre-endocrine
#> GCAGTTATCTTGAGAC GCAGTTATCTTGAGAC Ductal
#> GCATGATAGCCTTGAT GCATGATAGCCTTGAT Ngn3-high-EP
#> GCATGATCAAGCGAGT GCATGATCAAGCGAGT Endocrine
#> GCATGATGTCACTGGC GCATGATGTCACTGGC Endocrine
#> GCATGATGTTTAGGAA GCATGATGTTTAGGAA Endocrine
#> GCATGATTCAAACGGG GCATGATTCAAACGGG Ductal
#> GCATGCGAGGAACTGC GCATGCGAGGAACTGC Pre-endocrine
#> GCATGCGCAGCTCCGA GCATGCGCAGCTCCGA Endocrine
#> GCATGTAAGAATCTCC GCATGTAAGAATCTCC Endocrine
#> GCATGTAAGCGTGTCC GCATGTAAGCGTGTCC Endocrine
#> GCATGTAGTTGCGCAC GCATGTAGTTGCGCAC Endocrine
#> GCATGTATCGTGGACC GCATGTATCGTGGACC Endocrine
#> GCCTCTAAGCTAGTCT GCCTCTAAGCTAGTCT Endocrine
#> GCCTCTAAGTCCGGTC GCCTCTAAGTCCGGTC Ngn3-high-EP
#> GCCTCTATCGCTTAGA GCCTCTATCGCTTAGA Ngn3-low-EP
#> GCGACCATCACCAGGC GCGACCATCACCAGGC Endocrine
#> GCGAGAAGTGGGTATG GCGAGAAGTGGGTATG Endocrine
#> GCGCAACGTTTCGCTC GCGCAACGTTTCGCTC Endocrine
#> GCGCAACTCACAACGT GCGCAACTCACAACGT Ductal
#> GCGCAGTGTGCTAGCC GCGCAGTGTGCTAGCC Pre-endocrine
#> GCGCCAAAGCTCCTTC GCGCCAAAGCTCCTTC Endocrine
#> GCGCCAATCTTATCTG GCGCCAATCTTATCTG Pre-endocrine
#> GCGCGATGTCCGAATT GCGCGATGTCCGAATT Endocrine
#> GCGGGTTAGACTTGAA GCGGGTTAGACTTGAA Ngn3-high-EP
#> GCGGGTTCAAGTAATG GCGGGTTCAAGTAATG Endocrine
#> GCGGGTTGTTACTGAC GCGGGTTGTTACTGAC Ductal
#> GCTCCTAAGGGCATGT GCTCCTAAGGGCATGT Endocrine
#> GCTCTGTGTCCATGAT GCTCTGTGTCCATGAT Pre-endocrine
#> GCTGCAGGTTACCGAT GCTGCAGGTTACCGAT Ductal
#> GCTGCGAAGAACAACT GCTGCGAAGAACAACT Endocrine
#> GCTGCGAAGCTAGCCC GCTGCGAAGCTAGCCC Ngn3-low-EP
#> GCTGCGAAGTATTGGA GCTGCGAAGTATTGGA Ngn3-high-EP
#> GCTGCGATCCCGGATG GCTGCGATCCCGGATG Pre-endocrine
#> GCTGCGATCGGCTACG GCTGCGATCGGCTACG Endocrine
#> GCTGCTTGTCTTCAAG GCTGCTTGTCTTCAAG Ductal
#> GCTGGGTGTCATACTG GCTGGGTGTCATACTG Endocrine
#> GCTGGGTGTCTAGAGG GCTGGGTGTCTAGAGG Ngn3-high-EP
#> GCTTCCAAGACATAAC GCTTCCAAGACATAAC Endocrine
#> GCTTCCAAGTTAAGTG GCTTCCAAGTTAAGTG Endocrine
#> GCTTGAAGTCAACTGT GCTTGAAGTCAACTGT Endocrine
#> GGAAAGCCAATGTAAG GGAAAGCCAATGTAAG Pre-endocrine
#> GGAAAGCGTCTTGATG GGAAAGCGTCTTGATG Ductal
#> GGAATAAGTAAGAGAG GGAATAAGTAAGAGAG Endocrine
#> GGAATAAGTTCGGGCT GGAATAAGTTCGGGCT Ductal
#> GGACAAGGTGATAAGT GGACAAGGTGATAAGT Ductal
#> GGACAAGTCAAAGACA GGACAAGTCAAAGACA Ngn3-high-EP
#> GGACAGAAGGCCGAAT GGACAGAAGGCCGAAT Endocrine
#> GGACAGACAATCCAAC GGACAGACAATCCAAC Pre-endocrine
#> GGACAGATCAGGCGAA GGACAGATCAGGCGAA Endocrine
#> GGACAGATCCTGCAGG GGACAGATCCTGCAGG Ductal
#> GGACAGATCCTTTCGG GGACAGATCCTTTCGG Ngn3-high-EP
#> GGACATTGTCTCCCTA GGACATTGTCTCCCTA Endocrine
#> GGACGTCAGGGTGTTG GGACGTCAGGGTGTTG Pre-endocrine
#> GGACGTCCACGCATCG GGACGTCCACGCATCG Ngn3-low-EP
#> GGACGTCCATGGATGG GGACGTCCATGGATGG Ngn3-high-EP
#> GGACGTCGTCAGAAGC GGACGTCGTCAGAAGC Pre-endocrine
#> GGACGTCGTCGAGATG GGACGTCGTCGAGATG Endocrine
#> GGACGTCGTCTAGGTT GGACGTCGTCTAGGTT Endocrine
#> GGACGTCGTTTAGCTG GGACGTCGTTTAGCTG Ductal
#> GGAGCAAAGCTTCGCG GGAGCAAAGCTTCGCG Ductal
#> GGAGCAAGTATCACCA GGAGCAAGTATCACCA Pre-endocrine
#> GGATGTTAGTAAGTAC GGATGTTAGTAAGTAC Endocrine
#> GGATGTTTCCAGTATG GGATGTTTCCAGTATG Ngn3-high-EP
#> GGCAATTCACGGCTAC GGCAATTCACGGCTAC Endocrine
#> GGCAATTCATCTCCCA GGCAATTCATCTCCCA Ngn3-high-EP
#> GGCAATTGTAGAGCTG GGCAATTGTAGAGCTG Endocrine
#> GGCCGATTCACTCTTA GGCCGATTCACTCTTA Ngn3-low-EP
#> GGCGACTGTAAGTTCC GGCGACTGTAAGTTCC Endocrine
#> GGCGTGTCACCAGGTC GGCGTGTCACCAGGTC Ngn3-high-EP
#> GGCGTGTCACCGATAT GGCGTGTCACCGATAT Pre-endocrine
#> GGCGTGTTCAGCTTAG GGCGTGTTCAGCTTAG Endocrine
#> GGCGTGTTCGAGAGCA GGCGTGTTCGAGAGCA Pre-endocrine
#> GGCTCGAAGATCACGG GGCTCGAAGATCACGG Pre-endocrine
#> GGCTCGAAGTGACTCT GGCTCGAAGTGACTCT Endocrine
#> GGCTCGACAGCTTCGG GGCTCGACAGCTTCGG Endocrine
#> GGCTGGTAGTCACGCC GGCTGGTAGTCACGCC Ngn3-high-EP
#> GGCTGGTTCATGCATG GGCTGGTTCATGCATG Ngn3-high-EP
#> GGGAATGAGCACACAG GGGAATGAGCACACAG Endocrine
#> GGGAATGAGTGTACGG GGGAATGAGTGTACGG Ductal
#> GGGAATGCAAAGTGCG GGGAATGCAAAGTGCG Endocrine
#> GGGAATGTCACAAACC GGGAATGTCACAAACC Ngn3-high-EP
#> GGGACCTCACTGTCGG GGGACCTCACTGTCGG Pre-endocrine
#> GGGACCTTCGAATGCT GGGACCTTCGAATGCT Ductal
#> GGGAGATAGATGTGGC GGGAGATAGATGTGGC Endocrine
#> GGGAGATAGCGAAGGG GGGAGATAGCGAAGGG Endocrine
#> GGGAGATAGTGACATA GGGAGATAGTGACATA Endocrine
#> GGGATGAAGGGCATGT GGGATGAAGGGCATGT Endocrine
#> GGGATGAGTCTGCGGT GGGATGAGTCTGCGGT Endocrine
#> GGGATGAGTTGTGGAG GGGATGAGTTGTGGAG Ngn3-high-EP
#> GGGCACTCATTTCAGG GGGCACTCATTTCAGG Ductal
#> GGGCACTGTCTCCATC GGGCACTGTCTCCATC Pre-endocrine
#> GGGCACTGTGGCAAAC GGGCACTGTGGCAAAC Ductal
#> GGGCATCCATGACATC GGGCATCCATGACATC Ductal
#> GGGCATCCATTTCAGG GGGCATCCATTTCAGG Endocrine
#> GGGCATCGTGGTCTCG GGGCATCGTGGTCTCG Ngn3-high-EP
#> GGGTCTGCACAGGCCT GGGTCTGCACAGGCCT Ngn3-high-EP
#> GGGTCTGCATGAAGTA GGGTCTGCATGAAGTA Ngn3-high-EP
#> GGGTCTGTCATGTGGT GGGTCTGTCATGTGGT Pre-endocrine
#> GGGTCTGTCCATGAAC GGGTCTGTCCATGAAC Endocrine
#> GGGTTGCCAAGCTGAG GGGTTGCCAAGCTGAG Ngn3-low-EP
#> GGGTTGCTCGAACGGA GGGTTGCTCGAACGGA Ductal
#> GGTATTGGTACTCGCG GGTATTGGTACTCGCG Ngn3-high-EP
#> GGTATTGGTCCCGACA GGTATTGGTCCCGACA Ductal
#> GGTGTTAAGGATCGCA GGTGTTAAGGATCGCA Ductal
#> GGTGTTAGTTCCTCCA GGTGTTAGTTCCTCCA Ductal
#> GTAACGTGTCACTGGC GTAACGTGTCACTGGC Ductal
#> GTAACTGGTCACTGGC GTAACTGGTCACTGGC Pre-endocrine
#> GTAACTGTCAGTGCAT GTAACTGTCAGTGCAT Ductal
#> GTACGTAGTCTCTTTA GTACGTAGTCTCTTTA Endocrine
#> GTACTCCCACAGGTTT GTACTCCCACAGGTTT Pre-endocrine
#> GTACTCCCACGTCTCT GTACTCCCACGTCTCT Ngn3-low-EP
#> GTACTCCTCCAACCAA GTACTCCTCCAACCAA Ngn3-high-EP
#> GTACTTTGTACCGTAT GTACTTTGTACCGTAT Endocrine
#> GTACTTTTCTTCGGTC GTACTTTTCTTCGGTC Endocrine
#> GTAGGCCAGCTAACAA GTAGGCCAGCTAACAA Pre-endocrine
#> GTAGGCCGTTACCAGT GTAGGCCGTTACCAGT Endocrine
#> GTAGTCACATCGATGT GTAGTCACATCGATGT Ngn3-high-EP
#> GTAGTCAGTCCGACGT GTAGTCAGTCCGACGT Ngn3-high-EP
#> GTATCTTCAGTGGGAT GTATCTTCAGTGGGAT Ductal
#> GTATCTTCATCGATGT GTATCTTCATCGATGT Ngn3-high-EP
#> GTATCTTGTACTCAAC GTATCTTGTACTCAAC Endocrine
#> GTATCTTGTCGCGGTT GTATCTTGTCGCGGTT Ngn3-high-EP
#> GTATCTTGTGGCCCTA GTATCTTGTGGCCCTA Ngn3-high-EP
#> GTATCTTTCGTGGACC GTATCTTTCGTGGACC Pre-endocrine
#> GTATTCTCACATTAGC GTATTCTCACATTAGC Ductal
#> GTATTCTCACTTGGAT GTATTCTCACTTGGAT Ductal
#> GTATTCTGTCCGTCAG GTATTCTGTCCGTCAG Ngn3-low-EP
#> GTATTCTTCTCTAAGG GTATTCTTCTCTAAGG Endocrine
#> GTCAAGTGTGGTCCGT GTCAAGTGTGGTCCGT Ductal
#> GTCACAACACGTCTCT GTCACAACACGTCTCT Ngn3-high-EP
#> GTCACGGAGCTATGCT GTCACGGAGCTATGCT Endocrine
#> GTCACGGCATCCGTGG GTCACGGCATCCGTGG Pre-endocrine
#> GTCACGGTCTTGCCGT GTCACGGTCTTGCCGT Pre-endocrine
#> GTCATTTGTCGTCTTC GTCATTTGTCGTCTTC Endocrine
#> GTCCTCACACCACGTG GTCCTCACACCACGTG Endocrine
#> GTCCTCATCATTTGGG GTCCTCATCATTTGGG Ngn3-high-EP
#> GTCCTCATCTGGAGCC GTCCTCATCTGGAGCC Ngn3-high-EP
#> GTCGGGTCAGGCTGAA GTCGGGTCAGGCTGAA Endocrine
#> GTCGTAAAGTTTAGGA GTCGTAAAGTTTAGGA Endocrine
#> GTCTCGTAGGCTAGAC GTCTCGTAGGCTAGAC Ngn3-high-EP
#> GTCTCGTCATAGTAAG GTCTCGTCATAGTAAG Ngn3-high-EP
#> GTCTCGTTCACTTCAT GTCTCGTTCACTTCAT Ngn3-high-EP
#> GTCTCGTTCTGCGTAA GTCTCGTTCTGCGTAA Endocrine
#> GTCTTCGTCCGTTGCT GTCTTCGTCCGTTGCT Endocrine
#> GTCTTCGTCTACCAGA GTCTTCGTCTACCAGA Pre-endocrine
#> GTGCATAAGAACAATC GTGCATAAGAACAATC Pre-endocrine
#> GTGCATAAGCTCCTCT GTGCATAAGCTCCTCT Ductal
#> GTGCATAGTTCAACCA GTGCATAGTTCAACCA Endocrine
#> GTGCATATCAAGGCTT GTGCATATCAAGGCTT Endocrine
#> GTGCGGTAGTAGGCCA GTGCGGTAGTAGGCCA Ductal
#> GTGCGGTCAGCGTAAG GTGCGGTCAGCGTAAG Ngn3-high-EP
#> GTGCTTCAGAGGGATA GTGCTTCAGAGGGATA Endocrine
#> GTGCTTCTCACGACTA GTGCTTCTCACGACTA Endocrine
#> GTGGGTCCATAAAGGT GTGGGTCCATAAAGGT Endocrine
#> GTGGGTCCATCGATTG GTGGGTCCATCGATTG Endocrine
#> GTGGGTCGTCCAGTAT GTGGGTCGTCCAGTAT Endocrine
#> GTGGGTCTCCGAATGT GTGGGTCTCCGAATGT Endocrine
#> GTGTGCGGTGTAAGTA GTGTGCGGTGTAAGTA Endocrine
#> GTGTTAGCACCATCCT GTGTTAGCACCATCCT Ductal
#> GTGTTAGGTGAACCTT GTGTTAGGTGAACCTT Pre-endocrine
#> GTTAAGCAGGATTCGG GTTAAGCAGGATTCGG Ngn3-high-EP
#> GTTACAGTCCGATATG GTTACAGTCCGATATG Ngn3-high-EP
#> GTTCATTAGAAGATTC GTTCATTAGAAGATTC Ngn3-low-EP
#> GTTCATTAGGCAGTCA GTTCATTAGGCAGTCA Pre-endocrine
#> GTTCATTTCCGAACGC GTTCATTTCCGAACGC Endocrine
#> GTTCTCGAGAATTCCC GTTCTCGAGAATTCCC Endocrine
#> GTTCTCGCAAGAAAGG GTTCTCGCAAGAAAGG Endocrine
#> GTTCTCGCAGACACTT GTTCTCGCAGACACTT Ductal
#> GTTTCTAAGAAGATTC GTTTCTAAGAAGATTC Endocrine
#> GTTTCTACAGGACCCT GTTTCTACAGGACCCT Endocrine
#> TAAGAGATCTGCGACG TAAGAGATCTGCGACG Pre-endocrine
#> TAAGTGCAGGGTTTCT TAAGTGCAGGGTTTCT Ductal
#> TAAGTGCTCCAAATGC TAAGTGCTCCAAATGC Endocrine
#> TACACGACAACGATCT TACACGACAACGATCT Ductal
#> TACACGACATGTAGTC TACACGACATGTAGTC Endocrine
#> TACACGAGTGCAACTT TACACGAGTGCAACTT Ngn3-high-EP
#> TACAGTGCAATCTACG TACAGTGCAATCTACG Ngn3-low-EP
#> TACAGTGCACAGGCCT TACAGTGCACAGGCCT Endocrine
#> TACAGTGGTAAACGCG TACAGTGGTAAACGCG Pre-endocrine
#> TACCTTAAGTACATGA TACCTTAAGTACATGA Ductal
#> TACCTTATCCGATATG TACCTTATCCGATATG Endocrine
#> TACGGATAGAGCTGGT TACGGATAGAGCTGGT Ductal
#> TACGGATAGGCACATG TACGGATAGGCACATG Endocrine
#> TACGGATAGTTTAGGA TACGGATAGTTTAGGA Ngn3-low-EP
#> TACGGATGTGTTCGAT TACGGATGTGTTCGAT Ductal
#> TACGGATTCCAAGTAC TACGGATTCCAAGTAC Ngn3-high-EP
#> TACGGGCAGCCAACAG TACGGGCAGCCAACAG Endocrine
#> TACGGGCAGCGTCAAG TACGGGCAGCGTCAAG Ductal
#> TACGGGCTCTTCGGTC TACGGGCTCTTCGGTC Endocrine
#> TACGGTACATGTTCCC TACGGTACATGTTCCC Ductal
#> TACTCATAGGTGTTAA TACTCATAGGTGTTAA Pre-endocrine
#> TACTCATCAGATCCAT TACTCATCAGATCCAT Endocrine
#> TACTCGCAGTAAGTAC TACTCGCAGTAAGTAC Endocrine
#> TACTCGCCAGATCGGA TACTCGCCAGATCGGA Endocrine
#> TACTCGCGTCGGCATC TACTCGCGTCGGCATC Endocrine
#> TACTTACCAACTGCGC TACTTACCAACTGCGC Ductal
#> TACTTACCATGGGACA TACTTACCATGGGACA Ngn3-high-EP
#> TACTTGTAGACAAAGG TACTTGTAGACAAAGG Endocrine
#> TAGACCAAGGAATCGC TAGACCAAGGAATCGC Ductal
#> TAGAGCTGTATGAAAC TAGAGCTGTATGAAAC Ductal
#> TAGAGCTGTTGTCGCG TAGAGCTGTTGTCGCG Ductal
#> TAGCCGGTCAACACAC TAGCCGGTCAACACAC Ductal
#> TAGGCATTCCAAAGTC TAGGCATTCCAAAGTC Ngn3-low-EP
#> TAGTGGTCAACTGCGC TAGTGGTCAACTGCGC Ductal
#> TAGTGGTCATTCACTT TAGTGGTCATTCACTT Pre-endocrine
#> TAGTGGTTCCTCAACC TAGTGGTTCCTCAACC Pre-endocrine
#> TAGTTGGCACGTAAGG TAGTTGGCACGTAAGG Ngn3-high-EP
#> TAGTTGGCAGTAACGG TAGTTGGCAGTAACGG Pre-endocrine
#> TAGTTGGGTACCGTAT TAGTTGGGTACCGTAT Ductal
#> TAGTTGGTCTTGCAAG TAGTTGGTCTTGCAAG Endocrine
#> TATCAGGCACCAGATT TATCAGGCACCAGATT Ductal
#> TATCTCACAACTGCTA TATCTCACAACTGCTA Ductal
#> TATCTCACACGCGAAA TATCTCACACGCGAAA Endocrine
#> TATCTCACATTGTGCA TATCTCACATTGTGCA Endocrine
#> TATCTCAGTTCGCTAA TATCTCAGTTCGCTAA Ductal
#> TATGCCCAGAACAATC TATGCCCAGAACAATC Pre-endocrine
#> TATGCCCGTCTAGCCG TATGCCCGTCTAGCCG Ngn3-high-EP
#> TATGCCCTCTTACCTA TATGCCCTCTTACCTA Ngn3-low-EP
#> TATTACCTCGTTGCCT TATTACCTCGTTGCCT Endocrine
#> TCAACGATCAACCATG TCAACGATCAACCATG Pre-endocrine
#> TCAATCTCATCCGCGA TCAATCTCATCCGCGA Ngn3-high-EP
#> TCAATCTCATCTCCCA TCAATCTCATCTCCCA Ngn3-low-EP
#> TCAATCTGTCAAAGAT TCAATCTGTCAAAGAT Ngn3-low-EP
#> TCAATCTTCGAACGGA TCAATCTTCGAACGGA Endocrine
#> TCACAAGAGTGACATA TCACAAGAGTGACATA Endocrine
#> TCACAAGCAACGATGG TCACAAGCAACGATGG Endocrine
#> TCACAAGCAGACTCGC TCACAAGCAGACTCGC Endocrine
#> TCACGAAGTCCGAGTC TCACGAAGTCCGAGTC Ngn3-high-EP
#> TCACGAATCCTATGTT TCACGAATCCTATGTT Ngn3-low-EP
#> TCAGATGCACTCGACG TCAGATGCACTCGACG Pre-endocrine
#> TCAGATGGTACACCGC TCAGATGGTACACCGC Endocrine
#> TCAGCAACACCAGGCT TCAGCAACACCAGGCT Ductal
#> TCAGCAATCGCCGTGA TCAGCAATCGCCGTGA Pre-endocrine
#> TCAGCTCAGTCGTACT TCAGCTCAGTCGTACT Ngn3-low-EP
#> TCAGGTAAGGCGTACA TCAGGTAAGGCGTACA Ngn3-high-EP
#> TCAGGTAAGTCAATAG TCAGGTAAGTCAATAG Ductal
#> TCAGGTACAGTCACTA TCAGGTACAGTCACTA Endocrine
#> TCAGGTATCTGTCCGT TCAGGTATCTGTCCGT Endocrine
#> TCATTACGTCAGAAGC TCATTACGTCAGAAGC Ductal
#> TCATTTGCATTGGCGC TCATTTGCATTGGCGC Pre-endocrine
#> TCCACACCAGGCGATA TCCACACCAGGCGATA Endocrine
#> TCCCGATAGTCGTTTG TCCCGATAGTCGTTTG Pre-endocrine
#> TCCCGATCAACGATGG TCCCGATCAACGATGG Ngn3-high-EP
#> TCGAGGCAGAACAATC TCGAGGCAGAACAATC Endocrine
#> TCGAGGCAGGCATGGT TCGAGGCAGGCATGGT Pre-endocrine
#> TCGCGAGAGTACGCGA TCGCGAGAGTACGCGA Pre-endocrine
#> TCGCGAGAGTCCGTAT TCGCGAGAGTCCGTAT Endocrine
#> TCGCGAGCACTTCTGC TCGCGAGCACTTCTGC Pre-endocrine
#> TCGGGACAGAGAACAG TCGGGACAGAGAACAG Ngn3-high-EP
#> TCGGGACGTCTGCGGT TCGGGACGTCTGCGGT Endocrine
#> TCGTACCCACGCATCG TCGTACCCACGCATCG Ngn3-low-EP
#> TCGTACCCATATACGC TCGTACCCATATACGC Ductal
#> TCTATTGCACAGGAGT TCTATTGCACAGGAGT Ductal
#> TCTATTGTCCTATTCA TCTATTGTCCTATTCA Endocrine
#> TCTATTGTCGTATCAG TCTATTGTCGTATCAG Pre-endocrine
#> TCTCATAAGAATGTGT TCTCATAAGAATGTGT Pre-endocrine
#> TCTCATACAAATTGCC TCTCATACAAATTGCC Endocrine
#> TCTCATAGTGGACGAT TCTCATAGTGGACGAT Ngn3-high-EP
#> TCTCTAAAGCTGGAAC TCTCTAAAGCTGGAAC Endocrine
#> TCTCTAAGTCGCATCG TCTCTAAGTCGCATCG Ductal
#> TCTCTAAGTGGCAAAC TCTCTAAGTGGCAAAC Ngn3-high-EP
#> TCTCTAAGTTACCGAT TCTCTAAGTTACCGAT Ductal
#> TCTCTAATCCCAACGG TCTCTAATCCCAACGG Pre-endocrine
#> TCTGAGAGTCAAACTC TCTGAGAGTCAAACTC Ductal
#> TCTGGAACACACGCTG TCTGGAACACACGCTG Ductal
#> TCTGGAACACCTATCC TCTGGAACACCTATCC Endocrine
#> TCTGGAAGTCATCGGC TCTGGAAGTCATCGGC Endocrine
#> TCTGGAATCGGTGTCG TCTGGAATCGGTGTCG Ductal
#> TCTTTCCGTGCAACTT TCTTTCCGTGCAACTT Endocrine
#> TCTTTCCTCGTCTGCT TCTTTCCTCGTCTGCT Ngn3-low-EP
#> TGAAAGAGTTATCACG TGAAAGAGTTATCACG Ngn3-low-EP
#> TGACGGCAGGAATGGA TGACGGCAGGAATGGA Ductal
#> TGACGGCCATATGAGA TGACGGCCATATGAGA Endocrine
#> TGACTAGGTGTATGGG TGACTAGGTGTATGGG Ngn3-high-EP
#> TGACTAGTCCACGTGG TGACTAGTCCACGTGG Ngn3-low-EP
#> TGACTAGTCCTATTCA TGACTAGTCCTATTCA Ngn3-high-EP
#> TGACTTTAGCCAGAAC TGACTTTAGCCAGAAC Endocrine
#> TGACTTTAGCGTTGCC TGACTTTAGCGTTGCC Ductal
#> TGACTTTAGCGTTTAC TGACTTTAGCGTTTAC Ductal
#> TGAGAGGAGATCCCAT TGAGAGGAGATCCCAT Endocrine
#> TGAGAGGAGCCAGGAT TGAGAGGAGCCAGGAT Endocrine
#> TGAGAGGAGCCGTCGT TGAGAGGAGCCGTCGT Ngn3-high-EP
#> TGAGAGGAGGCGATAC TGAGAGGAGGCGATAC Endocrine
#> TGAGAGGAGGTAAACT TGAGAGGAGGTAAACT Endocrine
#> TGAGAGGCAGGAACGT TGAGAGGCAGGAACGT Endocrine
#> TGAGAGGGTAAGGGCT TGAGAGGGTAAGGGCT Ductal
#> TGAGCATGTTAAAGAC TGAGCATGTTAAAGAC Ngn3-high-EP
#> TGAGCATTCACCCGAG TGAGCATTCACCCGAG Ductal
#> TGAGCATTCGGGAGTA TGAGCATTCGGGAGTA Ductal
#> TGAGCCGTCATGTCCC TGAGCCGTCATGTCCC Ductal
#> TGAGCCGTCCGAACGC TGAGCCGTCCGAACGC Ductal
#> TGAGCCGTCCGCTGTT TGAGCCGTCCGCTGTT Ngn3-high-EP
#> TGAGGGAAGAGGTACC TGAGGGAAGAGGTACC Ngn3-high-EP
#> TGAGGGACAGACTCGC TGAGGGACAGACTCGC Ductal
#> TGATTTCAGTACGCCC TGATTTCAGTACGCCC Ngn3-high-EP
#> TGATTTCAGTGACTCT TGATTTCAGTGACTCT Ductal
#> TGATTTCGTGAGGGTT TGATTTCGTGAGGGTT Endocrine
#> TGCCAAACATGACATC TGCCAAACATGACATC Ngn3-high-EP
#> TGCCCATCACTGTGTA TGCCCATCACTGTGTA Ngn3-low-EP
#> TGCCCTAAGTCCGGTC TGCCCTAAGTCCGGTC Pre-endocrine
#> TGCGCAGGTACGCACC TGCGCAGGTACGCACC Ductal
#> TGCGCAGTCTGCCAGG TGCGCAGTCTGCCAGG Ngn3-low-EP
#> TGCGGGTAGACGCACA TGCGGGTAGACGCACA Ngn3-high-EP
#> TGCGGGTAGGGTTCCC TGCGGGTAGGGTTCCC Endocrine
#> TGCGGGTTCCACTCCA TGCGGGTTCCACTCCA Endocrine
#> TGCGGGTTCTCAAACG TGCGGGTTCTCAAACG Ductal
#> TGCGTGGAGGTAAACT TGCGTGGAGGTAAACT Ngn3-low-EP
#> TGCGTGGAGGTTCCTA TGCGTGGAGGTTCCTA Pre-endocrine
#> TGCGTGGAGTACACCT TGCGTGGAGTACACCT Endocrine
#> TGCGTGGGTCCCTTGT TGCGTGGGTCCCTTGT Pre-endocrine
#> TGCGTGGGTTACTGAC TGCGTGGGTTACTGAC Pre-endocrine
#> TGCTACCCAGGGCATA TGCTACCCAGGGCATA Ngn3-low-EP
#> TGCTACCCATAAGACA TGCTACCCATAAGACA Ngn3-high-EP
#> TGCTGCTAGGATGCGT TGCTGCTAGGATGCGT Ngn3-high-EP
#> TGCTGCTGTCCGTCAG TGCTGCTGTCCGTCAG Ductal
#> TGGACGCAGTGTTGAA TGGACGCAGTGTTGAA Ngn3-low-EP
#> TGGACGCTCAGGTAAA TGGACGCTCAGGTAAA Ductal
#> TGGACGCTCTTGTTTG TGGACGCTCTTGTTTG Pre-endocrine
#> TGGCCAGAGCTACCTA TGGCCAGAGCTACCTA Ductal
#> TGGCCAGCAAGGACTG TGGCCAGCAAGGACTG Endocrine
#> TGGCCAGCAATGAAAC TGGCCAGCAATGAAAC Pre-endocrine
#> TGGCCAGCACGAAAGC TGGCCAGCACGAAAGC Ductal
#> TGGCCAGGTCGGCATC TGGCCAGGTCGGCATC Ductal
#> TGGCGCATCACAACGT TGGCGCATCACAACGT Ductal
#> TGGCTGGGTACCCAAT TGGCTGGGTACCCAAT Endocrine
#> TGGGAAGCACGTAAGG TGGGAAGCACGTAAGG Pre-endocrine
#> TGGGAAGCATGACATC TGGGAAGCATGACATC Ductal
#> TGGGCGTGTGTGCGTC TGGGCGTGTGTGCGTC Endocrine
#> TGGTTAGCAGTCGTGC TGGTTAGCAGTCGTGC Pre-endocrine
#> TGGTTCCCACCGCTAG TGGTTCCCACCGCTAG Ductal
#> TGGTTCCGTCACCTAA TGGTTCCGTCACCTAA Ductal
#> TGTATTCCAATGACCT TGTATTCCAATGACCT Ductal
#> TGTCCCAAGTTTAGGA TGTCCCAAGTTTAGGA Ngn3-low-EP
#> TGTCCCACATTATCTC TGTCCCACATTATCTC Endocrine
#> TGTCCCAGTCCGTGAC TGTCCCAGTCCGTGAC Endocrine
#> TGTCCCATCTCTGCTG TGTCCCATCTCTGCTG Endocrine
#> TGTGGTACAAGACGTG TGTGGTACAAGACGTG Ductal
#> TGTGGTACACATCCGG TGTGGTACACATCCGG Endocrine
#> TGTGGTAGTGCCTGCA TGTGGTAGTGCCTGCA Endocrine
#> TGTGGTATCCAGTAGT TGTGGTATCCAGTAGT Ngn3-high-EP
#> TGTGTTTAGGAATGGA TGTGTTTAGGAATGGA Endocrine
#> TGTGTTTGTAGATTAG TGTGTTTGTAGATTAG Endocrine
#> TGTGTTTGTATGAATG TGTGTTTGTATGAATG Pre-endocrine
#> TTAACTCCAAAGAATC TTAACTCCAAAGAATC Ductal
#> TTAACTCCAGGATTGG TTAACTCCAGGATTGG Endocrine
#> TTAACTCGTAATCGTC TTAACTCGTAATCGTC Pre-endocrine
#> TTAGGCAGTTTGTTGG TTAGGCAGTTTGTTGG Pre-endocrine
#> TTATGCTCACCCAGTG TTATGCTCACCCAGTG Endocrine
#> TTCCCAGAGTTACCCA TTCCCAGAGTTACCCA Ngn3-high-EP
#> TTCCCAGGTATTCTCT TTCCCAGGTATTCTCT Ngn3-low-EP
#> TTCCCAGTCATAGCAC TTCCCAGTCATAGCAC Pre-endocrine
#> TTCGAAGAGTACGCCC TTCGAAGAGTACGCCC Ngn3-high-EP
#> TTCGGTCAGCTAGCCC TTCGGTCAGCTAGCCC Ngn3-high-EP
#> TTCGGTCAGGGCATGT TTCGGTCAGGGCATGT Endocrine
#> TTCGGTCGTATAGTAG TTCGGTCGTATAGTAG Endocrine
#> TTCGGTCTCGACCAGC TTCGGTCTCGACCAGC Pre-endocrine
#> TTCTACAAGATCGATA TTCTACAAGATCGATA Ngn3-low-EP
#> TTCTACACATAAAGGT TTCTACACATAAAGGT Pre-endocrine
#> TTCTACAGTACTCTCC TTCTACAGTACTCTCC Ngn3-low-EP
#> TTCTACATCGCTTAGA TTCTACATCGCTTAGA Endocrine
#> TTCTACATCTGCAGTA TTCTACATCTGCAGTA Ductal
#> TTCTCAAAGCCGCCTA TTCTCAAAGCCGCCTA Ductal
#> TTCTCAACAATAGAGT TTCTCAACAATAGAGT Ngn3-high-EP
#> TTCTCCTAGTCCATAC TTCTCCTAGTCCATAC Endocrine
#> TTCTCCTCAATGGTCT TTCTCCTCAATGGTCT Ductal
#> TTCTTAGTCAGTGTTG TTCTTAGTCAGTGTTG Endocrine
#> TTGAACGAGAGCAATT TTGAACGAGAGCAATT Endocrine
#> TTGAACGAGGAGTTTA TTGAACGAGGAGTTTA Ngn3-high-EP
#> TTGAACGGTGAGGGAG TTGAACGGTGAGGGAG Endocrine
#> TTGAACGGTGATGCCC TTGAACGGTGATGCCC Ductal
#> TTGACTTAGACAAGCC TTGACTTAGACAAGCC Ductal
#> TTGACTTGTTCCATGA TTGACTTGTTCCATGA Ngn3-high-EP
#> TTGACTTTCACCGGGT TTGACTTTCACCGGGT Pre-endocrine
#> TTGACTTTCAGCAACT TTGACTTTCAGCAACT Ductal
#> TTGACTTTCCAGTAGT TTGACTTTCCAGTAGT Ductal
#> TTGCCGTCACCGAATT TTGCCGTCACCGAATT Endocrine
#> TTGCCGTTCTCTGTCG TTGCCGTTCTCTGTCG Ductal
#> TTGCGTCCACCGATAT TTGCGTCCACCGATAT Ngn3-high-EP
#> TTGGAACAGAAGATTC TTGGAACAGAAGATTC Endocrine
#> TTGGAACGTATATGAG TTGGAACGTATATGAG Endocrine
#> TTGGCAAAGAGTAAGG TTGGCAAAGAGTAAGG Endocrine
#> TTGTAGGTCAAAGTAG TTGTAGGTCAAAGTAG Ngn3-high-EP
#> TTTACTGCAGCATACT TTTACTGCAGCATACT Ngn3-high-EP
#> TTTACTGGTGCTAGCC TTTACTGGTGCTAGCC Ductal
#> TTTATGCAGGTTACCT TTTATGCAGGTTACCT Endocrine
#> TTTATGCGTCGAACAG TTTATGCGTCGAACAG Ductal
#> TTTATGCTCCATTCTA TTTATGCTCCATTCTA Ductal
#> TTTCCTCCAGGATCGA TTTCCTCCAGGATCGA Endocrine
#> TTTCCTCGTGGCGAAT TTTCCTCGTGGCGAAT Pre-endocrine
#> TTTGCGCAGACAGACC TTTGCGCAGACAGACC Ductal
#> TTTGGTTAGACAGGCT TTTGGTTAGACAGGCT Endocrine
#> TTTGGTTCACCAGATT TTTGGTTCACCAGATT Ductal
#> TTTGGTTCACGAAGCA TTTGGTTCACGAAGCA Ngn3-high-EP
#> TTTGGTTTCCTTTCGG TTTGGTTTCCTTTCGG Endocrine
#> TTTGTCAAGAATGTGT TTTGTCAAGAATGTGT Pre-endocrine
#> TTTGTCATCGAATGCT TTTGTCATCGAATGCT Endocrine
#> TTTGTCATCTGTTTGT TTTGTCATCTGTTTGT Endocrine
#>
#> $feature_metadata
#> features features_uique duplicated feature_split index
#> AUCell_A AUCell_A AUCell_A FALSE NA 1
#> GSVA_A GSVA_A GSVA_A FALSE NA 2
#> Sox9 Sox9 Sox9 FALSE NA 3
#> Anxa2 Anxa2 Anxa2 FALSE NA 4
#> Bicc1 Bicc1 Bicc1 FALSE NA 5
#>
#> $enrichment
#> NULL
#>