Single-cell reference mapping with Seurat method
Usage
RunSeuratMap(
srt_query,
srt_ref,
query_assay = NULL,
ref_pca = NULL,
ref_assay = srt_ref[[ref_pca]]@assay.used,
ref_dims = 1:30,
ref_umap = NULL,
ref_group = NULL,
normalization.method = "LogNormalize",
reduction_project_method = "pcaproject",
k.anchor = 5,
k.filter = 200,
k.score = 30,
k.weight = 100,
projection_method = c("model", "knn"),
nn_method = NULL,
k = 30,
distance_metric = "cosine",
vote_fun = "mean",
verbose = TRUE
)Arguments
- srt_query
An object of class Seurat to be annotated with cell types.
- srt_ref
A Seurat object or count matrix representing the reference object. If provided, the similarities will be calculated between cells from the query and reference objects. If not provided, the similarities will be calculated within the query object.
- query_assay
The assay to use for the query object. If not provided, the default assay of the query object will be used.
- ref_pca
A character string specifying the name of the PCA reduction in the reference object to use for calculating the distance metric.
- ref_assay
The assay to use for the reference object. If not provided, the default assay of the reference object will be used.
- ref_dims
A numeric vector specifying the dimension indices from the reference reduction to be used for calculating the distance metric.
- ref_umap
A character string specifying the name of the UMAP reduction in the reference object. If not provided, the first UMAP reduction found in the reference object will be used.
- ref_group
The grouping variable in the reference object. This variable will be used to group cells in the heatmap columns. If not provided, all cells will be treated as one group.
- normalization.method
The normalization method to use. Default is `"LogNormalize"`.
- reduction_project_method
Dimensional reduction to perform when finding anchors. Default is `"pcaproject"`.
- k.anchor
How many neighbors (k) to use when finding anchors. Default is `5`.
- k.filter
How many neighbors (k) to use when filtering anchors. Set to NA to turn off filtering. Default is `200`.
- k.score
How many neighbors (k) to use when scoring anchors. Default is `30`.
- k.weight
Number of neighbors to consider when weighting anchors. Default is `100`.
- projection_method
A character string specifying the projection method to use. Options are "model" and "knn". If "model" is selected, the function will try to use a pre-trained UMAP model in the reference object for projection. If "knn" is selected, the function will directly find the nearest neighbors using the distance metric.
- nn_method
A character string specifying the nearest neighbor search method to use. Options are "raw", "annoy", and "rann". If "raw" is selected, the function will use the brute-force method to find the nearest neighbors. If "annoy" is selected, the function will use the Annoy library for approximate nearest neighbor search. If "rann" is selected, the function will use the RANN library for approximate nearest neighbor search. If not provided, the function will choose the search method based on the size of the query and reference datasets.
- k
A number of nearest neighbors to find for each cell in the query object.
- distance_metric
The distance metric to use for calculating the pairwise distances between cells. Options include: "pearson", "spearman", "cosine", "correlation", "jaccard", "ejaccard", "dice", "edice", "hamman", "simple matching", and "faith". Additional distance metrics can also be used, such as "euclidean", "manhattan", "hamming", etc.
- vote_fun
A character string specifying the function to be used for aggregating the nearest neighbors in the reference object. Options are "mean", "median", "sum", "min", "max", "sd", "var", etc. If not provided, the default is "mean".
- verbose
Whether to print the message. Default is
TRUE.
Examples
data(panc8_sub)
panc8_sub <- standard_scop(panc8_sub)
#> ℹ [2026-07-02 09:52:02] Start standard processing workflow...
#> ℹ [2026-07-02 09:52:03] Checking a list of <Seurat>...
#> ! [2026-07-02 09:52:03] Data 1/1 of the `srt_list` is "unknown"
#> ℹ [2026-07-02 09:52:03] Perform `NormalizeData()` with `normalization.method = 'LogNormalize'` on 1/1 of `srt_list`...
#> ℹ [2026-07-02 09:52:03] Perform `FindVariableFeatures()` on 1/1 of `srt_list`...
#> ℹ [2026-07-02 09:52:03] Use the separate HVF from `srt_list`
#> ℹ [2026-07-02 09:52:03] Number of available HVF: 2000
#> ℹ [2026-07-02 09:52:03] Finished check
#> ℹ [2026-07-02 09:52:03] Perform `ScaleData()`
#> ℹ [2026-07-02 09:52:04] Perform pca linear dimension reduction
#> ℹ [2026-07-02 09:52:05] Use stored estimated dimensions 1:27 for Standardpca
#> ℹ [2026-07-02 09:52:05] Perform `Seurat::FindClusters()` with `cluster_algorithm = 'louvain'` and `cluster_resolution = 0.6`
#> ℹ [2026-07-02 09:52:05] Reorder clusters...
#> ℹ [2026-07-02 09:52:05] Skip `log1p()` because `layer = data` is not "counts"
#> ℹ [2026-07-02 09:52:05] Perform umap nonlinear dimension reduction
#> ✔ [2026-07-02 09:52:13] Standard processing workflow completed
srt_ref <- panc8_sub[, panc8_sub$tech != "fluidigmc1"]
srt_query <- panc8_sub[, panc8_sub$tech == "fluidigmc1"]
srt_ref <- integration_scop(
srt_ref,
batch = "tech",
integration_method = "Uncorrected"
)
#> ◌ [2026-07-02 09:52:13] Run integration workflow...
#> ℹ [2026-07-02 09:52:14] Split `srt_merge` into `srt_list` by "tech"
#> ℹ [2026-07-02 09:52:14] Checking a list of <Seurat>...
#> ℹ [2026-07-02 09:52:15] Data 1/4 of the `srt_list` has been log-normalized
#> ℹ [2026-07-02 09:52:15] Perform `FindVariableFeatures()` on 1/4 of `srt_list`...
#> ℹ [2026-07-02 09:52:15] Data 2/4 of the `srt_list` has been log-normalized
#> ℹ [2026-07-02 09:52:15] Perform `FindVariableFeatures()` on 2/4 of `srt_list`...
#> ℹ [2026-07-02 09:52:15] Data 3/4 of the `srt_list` has been log-normalized
#> ℹ [2026-07-02 09:52:15] Perform `FindVariableFeatures()` on 3/4 of `srt_list`...
#> ℹ [2026-07-02 09:52:16] Data 4/4 of the `srt_list` has been log-normalized
#> ℹ [2026-07-02 09:52:16] Perform `FindVariableFeatures()` on 4/4 of `srt_list`...
#> ℹ [2026-07-02 09:52:16] Use the separate HVF from `srt_list`
#> ℹ [2026-07-02 09:52:16] Number of available HVF: 2000
#> ℹ [2026-07-02 09:52:17] Finished check
#> ℹ [2026-07-02 09:52:17] Perform Uncorrected integration
#> ℹ [2026-07-02 09:52:17] Perform `Seurat::ScaleData()`
#> ℹ [2026-07-02 09:52:17] Perform "pca" linear dimension reduction
#> ! [2026-07-02 09:52:17] Some PCA features are absent from scale.data and will be dropped: "G0S2", "MRC2", "COL18A1", "COL5A3", "NOTCH3", "CRLF1", "PTGR1", "IL11", "LIF", "PDLIM3", "HTRA1", "TFPI2", "NREP", "ENG", "AQP3", "SEMA7A", "NPTX2", "SNAI2", …, "PCDH18", and "SCN11A"
#> ℹ [2026-07-02 09:52:17] Adjust neighbor k from 20 to 20 for small-sample clustering
#> ℹ [2026-07-02 09:52:17] Perform `Seurat::FindClusters()` with "louvain"
#> ℹ [2026-07-02 09:52:17] Reorder clusters...
#> ℹ [2026-07-02 09:52:18] Skip `log1p()` because `layer = data` is not "counts"
#> ℹ [2026-07-02 09:52:18] Perform umap nonlinear dimension reduction using Uncorrectedpca (1:25)
#> ℹ [2026-07-02 09:52:23] Perform umap nonlinear dimension reduction using Uncorrectedpca (1:25)
#> ℹ [2026-07-02 09:52:29] Perform umap nonlinear dimension reduction using Uncorrectedpca (1:25)
#> ✔ [2026-07-02 09:52:34] Uncorrected integration completed
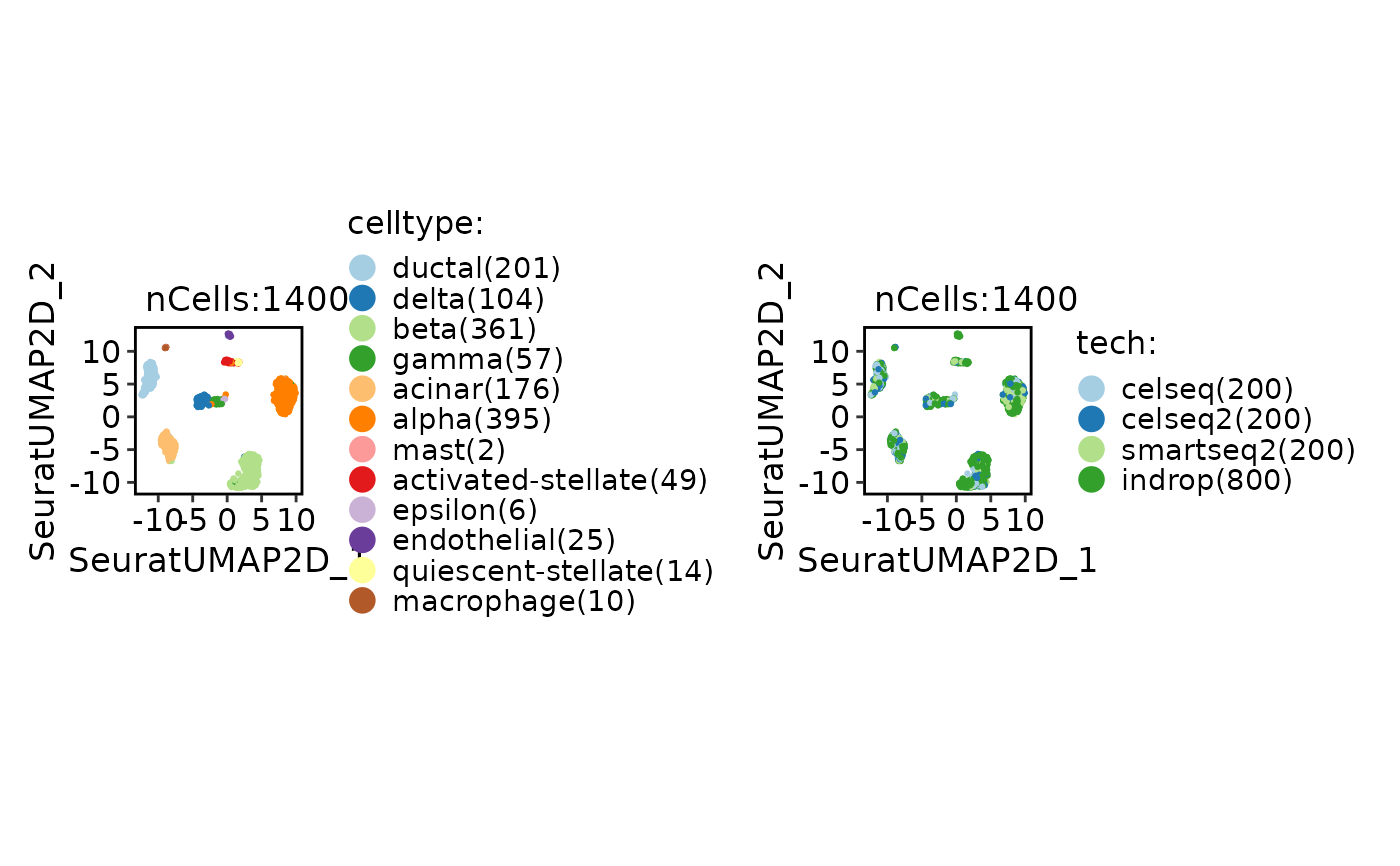
CellDimPlot(srt_ref, group.by = c("celltype", "tech"))
 # Projection
srt_query <- RunSeuratMap(
srt_query = srt_query,
srt_ref = srt_ref,
ref_pca = "Uncorrectedpca",
ref_umap = "UncorrectedUMAP2D",
k.weight = 50
)
#> ℹ [2026-07-02 09:52:35] Data type is log-normalized
#> ℹ [2026-07-02 09:52:35] Detected srt_query data type: log_normalized_counts
#> ℹ [2026-07-02 09:52:36] Data type is log-normalized
#> ℹ [2026-07-02 09:52:36] Detected srt_ref data type: log_normalized_counts
#> ℹ [2026-07-02 09:52:36] Run FindTransferAnchors
#> Projecting cell embeddings
#> Finding neighborhoods
#> Finding anchors
#> Found 474 anchors
#> Filtering anchors
#> Retained 474 anchors
#> Requested to reuse weights matrix, but no weights found. Computing new weights.
#> Warning: Layer counts isn't present in the assay object; returning NULL
#> Warning: Layer counts isn't present in the assay object; returning NULL
#>
#> Integrating dataset 2 with reference dataset
#> Finding integration vectors
#> Finding integration vector weights
#> Integrating data
#> ℹ [2026-07-02 09:52:41] Run UMAP projection
#> ℹ [2026-07-02 09:52:41] Use the reduction to calculate distance metric
#> ℹ [2026-07-02 09:52:41] Use raw method to find neighbors
#> ℹ [2026-07-02 09:52:41] Running UMAP projection
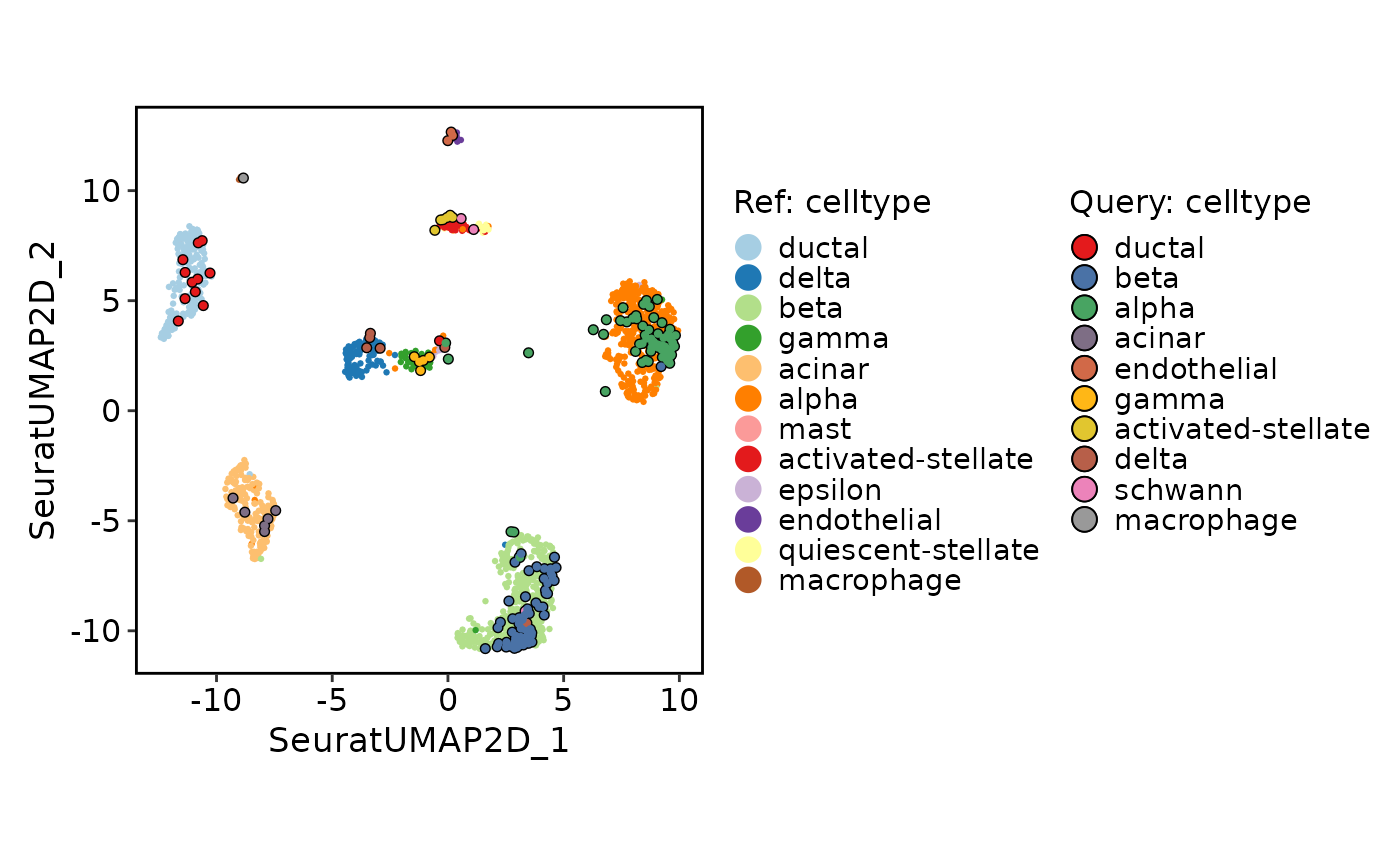
ProjectionPlot(
srt_query = srt_query,
srt_ref = srt_ref,
query_group = "celltype",
ref_group = "celltype"
)
#> Scale for x is already present.
#> Adding another scale for x, which will replace the existing scale.
#> Scale for y is already present.
#> Adding another scale for y, which will replace the existing scale.
#> Warning: Removed 1 row containing missing values or values outside the scale range
#> (`geom_point()`).
#> Warning: Removed 1 row containing missing values or values outside the scale range
#> (`geom_point()`).
# Projection
srt_query <- RunSeuratMap(
srt_query = srt_query,
srt_ref = srt_ref,
ref_pca = "Uncorrectedpca",
ref_umap = "UncorrectedUMAP2D",
k.weight = 50
)
#> ℹ [2026-07-02 09:52:35] Data type is log-normalized
#> ℹ [2026-07-02 09:52:35] Detected srt_query data type: log_normalized_counts
#> ℹ [2026-07-02 09:52:36] Data type is log-normalized
#> ℹ [2026-07-02 09:52:36] Detected srt_ref data type: log_normalized_counts
#> ℹ [2026-07-02 09:52:36] Run FindTransferAnchors
#> Projecting cell embeddings
#> Finding neighborhoods
#> Finding anchors
#> Found 474 anchors
#> Filtering anchors
#> Retained 474 anchors
#> Requested to reuse weights matrix, but no weights found. Computing new weights.
#> Warning: Layer counts isn't present in the assay object; returning NULL
#> Warning: Layer counts isn't present in the assay object; returning NULL
#>
#> Integrating dataset 2 with reference dataset
#> Finding integration vectors
#> Finding integration vector weights
#> Integrating data
#> ℹ [2026-07-02 09:52:41] Run UMAP projection
#> ℹ [2026-07-02 09:52:41] Use the reduction to calculate distance metric
#> ℹ [2026-07-02 09:52:41] Use raw method to find neighbors
#> ℹ [2026-07-02 09:52:41] Running UMAP projection
ProjectionPlot(
srt_query = srt_query,
srt_ref = srt_ref,
query_group = "celltype",
ref_group = "celltype"
)
#> Scale for x is already present.
#> Adding another scale for x, which will replace the existing scale.
#> Scale for y is already present.
#> Adding another scale for y, which will replace the existing scale.
#> Warning: Removed 1 row containing missing values or values outside the scale range
#> (`geom_point()`).
#> Warning: Removed 1 row containing missing values or values outside the scale range
#> (`geom_point()`).