Run doublet-calling with scds
Arguments
- srt
A Seurat object.
- assay
The name of the assay to be used for doublet-calling. Default is
"RNA".- db_rate
The expected doublet rate. Default is calculated as
ncol(srt) / 1000 * 0.01.- method
The method to be used for doublet-calling. Options are
"hybrid","cxds", or"bcds".- data_type
Optional precomputed result from CheckDataType for the input assay. Primarily used internally to avoid repeated scans of the same count matrix across nested QC calls.
- ...
Additional arguments passed to the selected
scdsmethod.- verbose
Whether to print the message. Default is
TRUE.
Examples
data(pancreas_sub)
pancreas_sub <- standard_scop(pancreas_sub)
#> ℹ [2026-07-02 10:13:46] Start standard processing workflow...
#> ℹ [2026-07-02 10:13:47] Checking a list of <Seurat>...
#> ! [2026-07-02 10:13:47] Data 1/1 of the `srt_list` is "unknown"
#> ℹ [2026-07-02 10:13:47] Perform `NormalizeData()` with `normalization.method = 'LogNormalize'` on 1/1 of `srt_list`...
#> ℹ [2026-07-02 10:13:47] Perform `FindVariableFeatures()` on 1/1 of `srt_list`...
#> ℹ [2026-07-02 10:13:47] Use the separate HVF from `srt_list`
#> ℹ [2026-07-02 10:13:47] Number of available HVF: 2000
#> ℹ [2026-07-02 10:13:47] Finished check
#> ℹ [2026-07-02 10:13:47] Perform `ScaleData()`
#> ℹ [2026-07-02 10:13:47] Perform pca linear dimension reduction
#> ℹ [2026-07-02 10:13:48] Use stored estimated dimensions 1:23 for Standardpca
#> ℹ [2026-07-02 10:13:48] Perform `Seurat::FindClusters()` with `cluster_algorithm = 'louvain'` and `cluster_resolution = 0.6`
#> ℹ [2026-07-02 10:13:48] Reorder clusters...
#> ℹ [2026-07-02 10:13:49] Skip `log1p()` because `layer = data` is not "counts"
#> ℹ [2026-07-02 10:13:49] Perform umap nonlinear dimension reduction
#> ✔ [2026-07-02 10:13:55] Standard processing workflow completed
pancreas_sub <- db_scds(pancreas_sub, method = "hybrid")
#> ℹ [2026-07-02 10:13:55] Running scds with method "hybrid"
#> ℹ [2026-07-02 10:13:56] Data type is raw counts
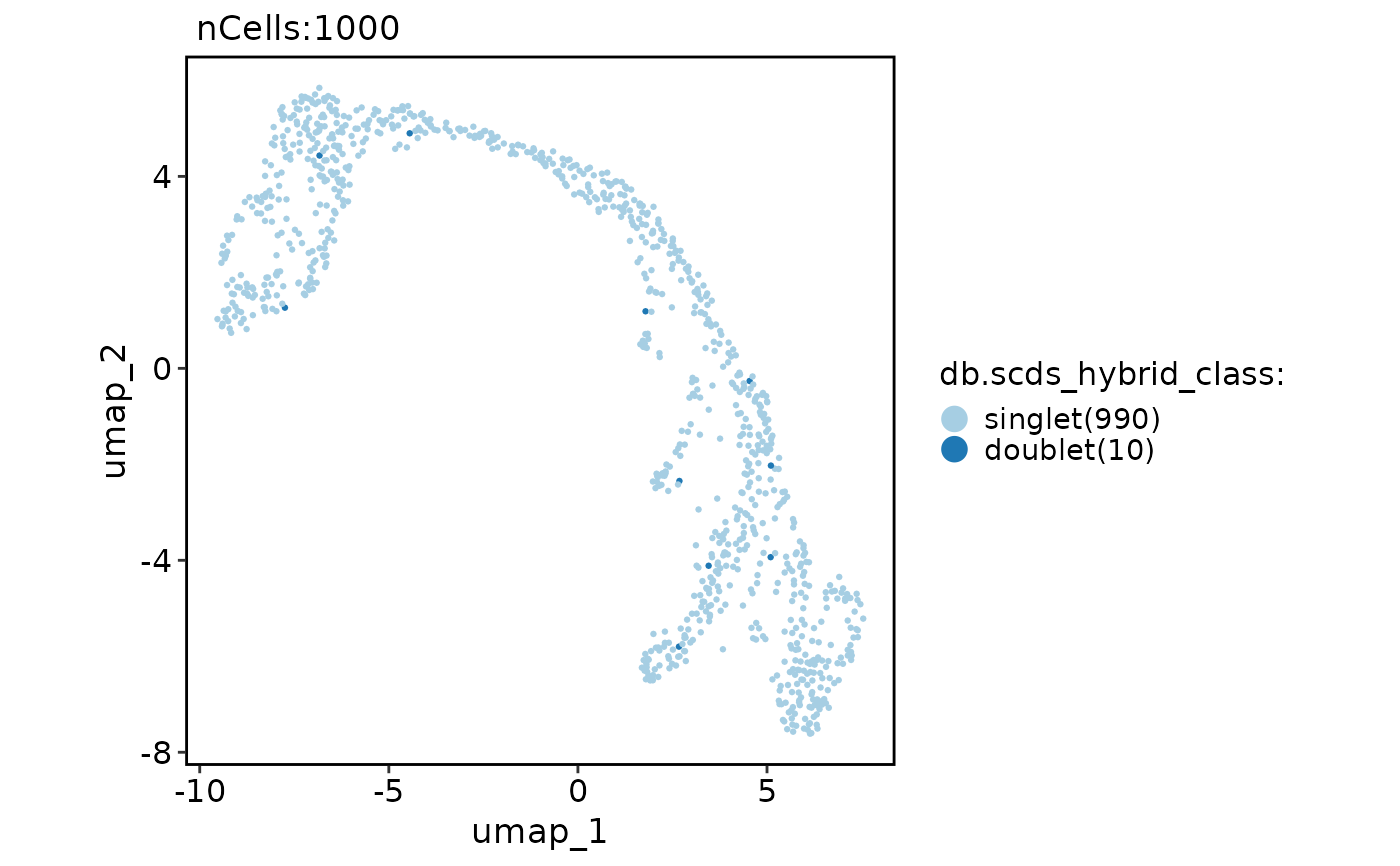
CellDimPlot(
pancreas_sub,
reduction = "umap",
group.by = "db.scds_hybrid_class"
)
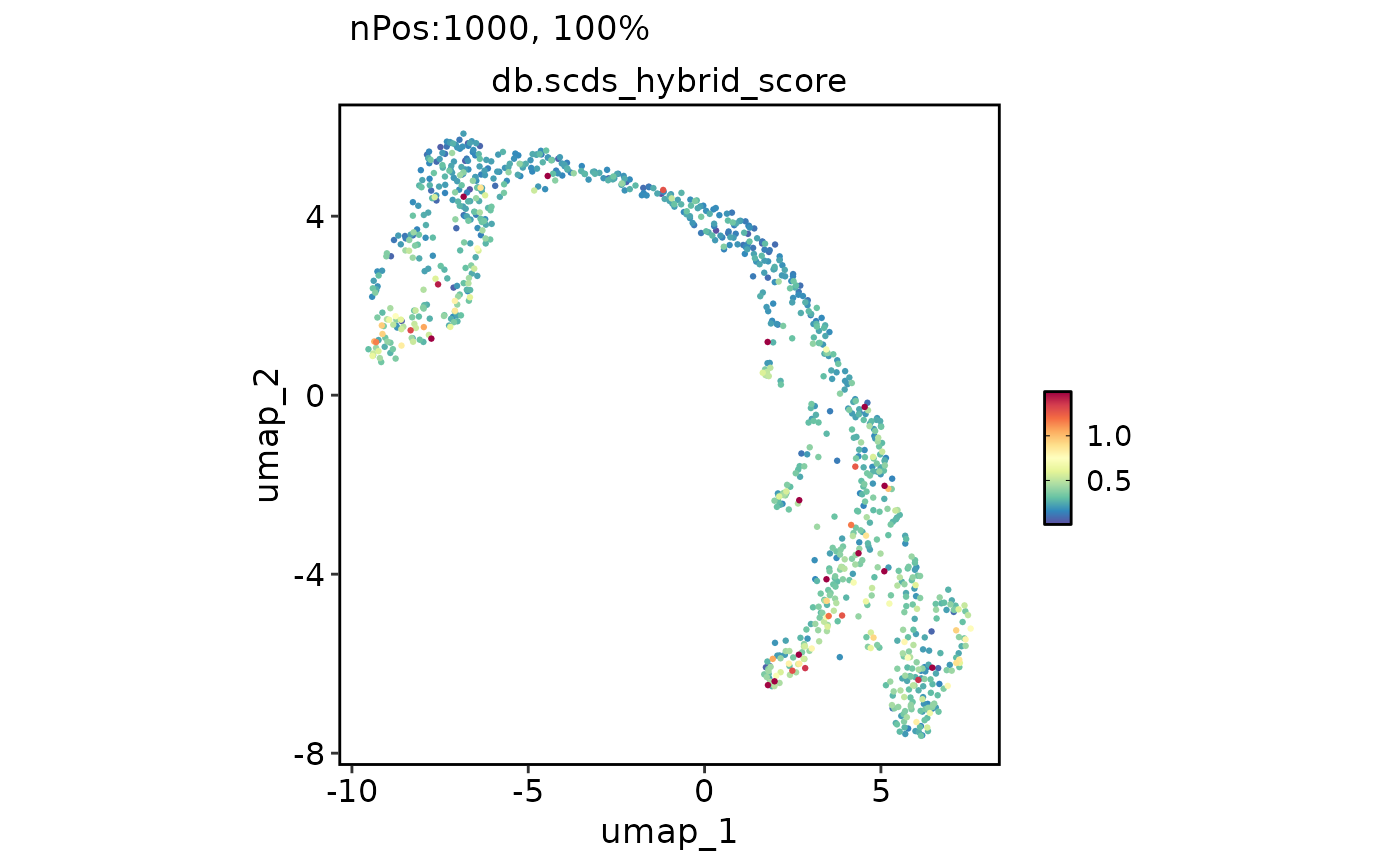
 FeatureDimPlot(
pancreas_sub,
reduction = "umap",
features = "db.scds_hybrid_score"
)
FeatureDimPlot(
pancreas_sub,
reduction = "umap",
features = "db.scds_hybrid_score"
)