Annotate single cells using scmap.
Usage
RunScmap(
srt_query,
srt_ref,
ref_group = NULL,
query_assay = "RNA",
ref_assay = "RNA",
method = "scmapCluster",
nfeatures = 500,
threshold = 0.5,
k = 10,
verbose = TRUE
)Arguments
- srt_query
An object of class Seurat to be annotated with cell types.
- srt_ref
An object of class Seurat storing the reference cells.
- ref_group
A character vector specifying the column name in the
srt_refmetadata that represents the cell grouping.- query_assay
A character vector specifying the assay to be used for the query data. Default is the default assay of the
srt_queryobject.- ref_assay
A character vector specifying the assay to be used for the reference data. Default is the default assay of the
srt_refobject.- method
The method to be used for scmap analysis. Can be any of
"scmapCluster"or"scmapCell". Default is"scmapCluster".- nfeatures
The number of top features to be selected. Default is
500.- threshold
The threshold value on similarity to determine if a cell is assigned to a cluster. This should be a value between
0and1. Default is0.5.- k
Number of clusters per group for k-means clustering when
methodis"scmapCell". Default is10.- verbose
Whether to print the message. Default is
TRUE.
Examples
data(panc8_sub)
panc8_sub <- standard_scop(panc8_sub)
#> ℹ [2026-07-02 09:50:49] Start standard processing workflow...
#> ℹ [2026-07-02 09:50:49] Checking a list of <Seurat>...
#> ! [2026-07-02 09:50:49] Data 1/1 of the `srt_list` is "unknown"
#> ℹ [2026-07-02 09:50:49] Perform `NormalizeData()` with `normalization.method = 'LogNormalize'` on 1/1 of `srt_list`...
#> ℹ [2026-07-02 09:50:50] Perform `FindVariableFeatures()` on 1/1 of `srt_list`...
#> ℹ [2026-07-02 09:50:50] Use the separate HVF from `srt_list`
#> ℹ [2026-07-02 09:50:50] Number of available HVF: 2000
#> ℹ [2026-07-02 09:50:50] Finished check
#> ℹ [2026-07-02 09:50:50] Perform `ScaleData()`
#> ℹ [2026-07-02 09:50:50] Perform pca linear dimension reduction
#> ℹ [2026-07-02 09:50:51] Use stored estimated dimensions 1:27 for Standardpca
#> ℹ [2026-07-02 09:50:52] Perform `Seurat::FindClusters()` with `cluster_algorithm = 'louvain'` and `cluster_resolution = 0.6`
#> ℹ [2026-07-02 09:50:52] Reorder clusters...
#> ℹ [2026-07-02 09:50:52] Skip `log1p()` because `layer = data` is not "counts"
#> ℹ [2026-07-02 09:50:52] Perform umap nonlinear dimension reduction
#> ✔ [2026-07-02 09:50:59] Standard processing workflow completed
genenames <- make.unique(
thisutils::capitalize(
rownames(panc8_sub),
force_tolower = TRUE
)
)
names(genenames) <- rownames(panc8_sub)
panc8_sub <- RenameFeatures(
panc8_sub,
newnames = genenames
)
#> ℹ [2026-07-02 09:50:59] Rename features for the assay: RNA
panc8_sub <- CheckDataMerge(
panc8_sub,
batch = "tech"
)[["srt_merge"]]
#> ℹ [2026-07-02 09:51:00] Split `srt_merge` into `srt_list` by "tech"
#> ℹ [2026-07-02 09:51:01] Checking a list of <Seurat>...
#> ℹ [2026-07-02 09:51:01] Data 1/5 of the `srt_list` has been log-normalized
#> ℹ [2026-07-02 09:51:01] Perform `FindVariableFeatures()` on 1/5 of `srt_list`...
#> ℹ [2026-07-02 09:51:02] Data 2/5 of the `srt_list` has been log-normalized
#> ℹ [2026-07-02 09:51:02] Perform `FindVariableFeatures()` on 2/5 of `srt_list`...
#> ℹ [2026-07-02 09:51:02] Data 3/5 of the `srt_list` has been log-normalized
#> ℹ [2026-07-02 09:51:02] Perform `FindVariableFeatures()` on 3/5 of `srt_list`...
#> ℹ [2026-07-02 09:51:02] Data 4/5 of the `srt_list` has been log-normalized
#> ℹ [2026-07-02 09:51:02] Perform `FindVariableFeatures()` on 4/5 of `srt_list`...
#> ℹ [2026-07-02 09:51:02] Data 5/5 of the `srt_list` has been log-normalized
#> ℹ [2026-07-02 09:51:02] Perform `FindVariableFeatures()` on 5/5 of `srt_list`...
#> ℹ [2026-07-02 09:51:03] Use the separate HVF from `srt_list`
#> ℹ [2026-07-02 09:51:03] Number of available HVF: 2000
#> ℹ [2026-07-02 09:51:03] Finished check
#> Warning: Key ‘StandardpcaUMAP2D_’ taken, using ‘standardpcaumap2d_’ instead
data(pancreas_sub)
pancreas_sub <- standard_scop(pancreas_sub)
#> ℹ [2026-07-02 09:51:03] Start standard processing workflow...
#> ℹ [2026-07-02 09:51:04] Checking a list of <Seurat>...
#> ! [2026-07-02 09:51:04] Data 1/1 of the `srt_list` is "unknown"
#> ℹ [2026-07-02 09:51:04] Perform `NormalizeData()` with `normalization.method = 'LogNormalize'` on 1/1 of `srt_list`...
#> ℹ [2026-07-02 09:51:04] Perform `FindVariableFeatures()` on 1/1 of `srt_list`...
#> ℹ [2026-07-02 09:51:04] Use the separate HVF from `srt_list`
#> ℹ [2026-07-02 09:51:05] Number of available HVF: 2000
#> ℹ [2026-07-02 09:51:05] Finished check
#> ℹ [2026-07-02 09:51:05] Perform `ScaleData()`
#> ℹ [2026-07-02 09:51:05] Perform pca linear dimension reduction
#> ℹ [2026-07-02 09:51:06] Use stored estimated dimensions 1:23 for Standardpca
#> ℹ [2026-07-02 09:51:06] Perform `Seurat::FindClusters()` with `cluster_algorithm = 'louvain'` and `cluster_resolution = 0.6`
#> ℹ [2026-07-02 09:51:06] Reorder clusters...
#> ℹ [2026-07-02 09:51:06] Skip `log1p()` because `layer = data` is not "counts"
#> ℹ [2026-07-02 09:51:06] Perform umap nonlinear dimension reduction
#> ✔ [2026-07-02 09:51:13] Standard processing workflow completed
pancreas_sub <- RunScmap(
srt_query = pancreas_sub,
srt_ref = panc8_sub,
ref_group = "celltype",
method = "scmapCluster"
)
#> ℹ [2026-07-02 09:51:46] Data type is log-normalized
#> ℹ [2026-07-02 09:51:46] Detected `srt_query` data type: "log_normalized_counts"
#> ℹ [2026-07-02 09:51:46] Data type is log-normalized
#> ℹ [2026-07-02 09:51:46] Detected `srt_ref` data type: "log_normalized_counts"
#> ℹ [2026-07-02 09:51:47] Perform selectFeatures
#> ℹ [2026-07-02 09:51:48] Perform indexCluster
#> ℹ [2026-07-02 09:51:48] Perform scmapCluster
#> Warning: Features Mt-atp6, Mt-co1, Mt-co2, Mt-co3, Mt-nd1, Mt-nd2, Mt-nd4, Mt-nd4l, Mt-nd5 are not present in the 'SCESet' object and therefore were not set.
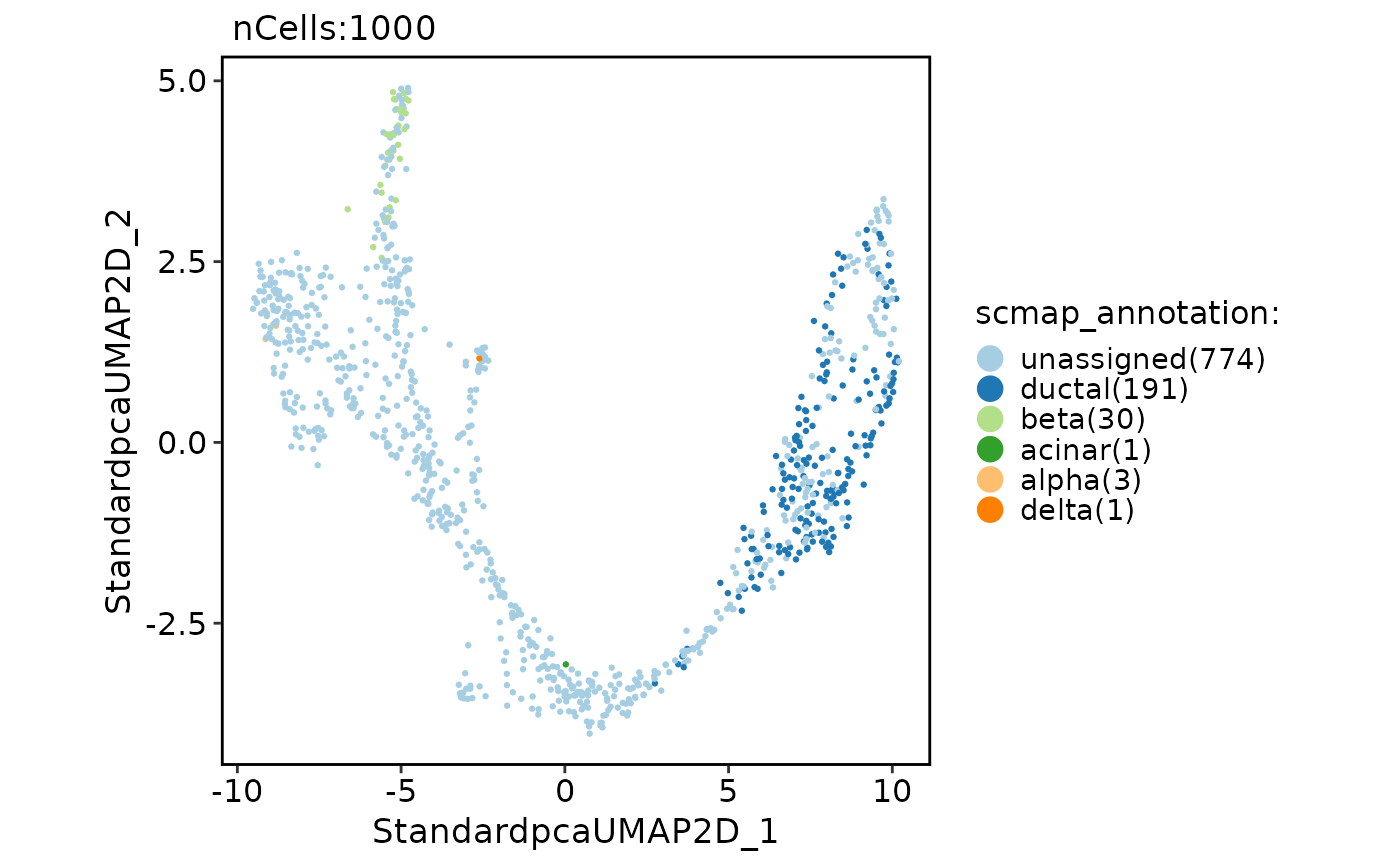
CellDimPlot(
pancreas_sub,
group.by = "scmap_annotation"
)
 pancreas_sub <- RunScmap(
srt_query = pancreas_sub,
srt_ref = panc8_sub,
ref_group = "celltype",
method = "scmapCell"
)
#> ℹ [2026-07-02 09:51:51] Data type is log-normalized
#> ℹ [2026-07-02 09:51:51] Detected `srt_query` data type: "log_normalized_counts"
#> ℹ [2026-07-02 09:51:52] Data type is log-normalized
#> ℹ [2026-07-02 09:51:52] Detected `srt_ref` data type: "log_normalized_counts"
#> ℹ [2026-07-02 09:51:53] Perform selectFeatures
#> ℹ [2026-07-02 09:51:53] Perform indexCell
#> ℹ [2026-07-02 09:51:53] Perform scmapCell
#> ℹ [2026-07-02 09:51:54] Perform scmapCell2Cluster
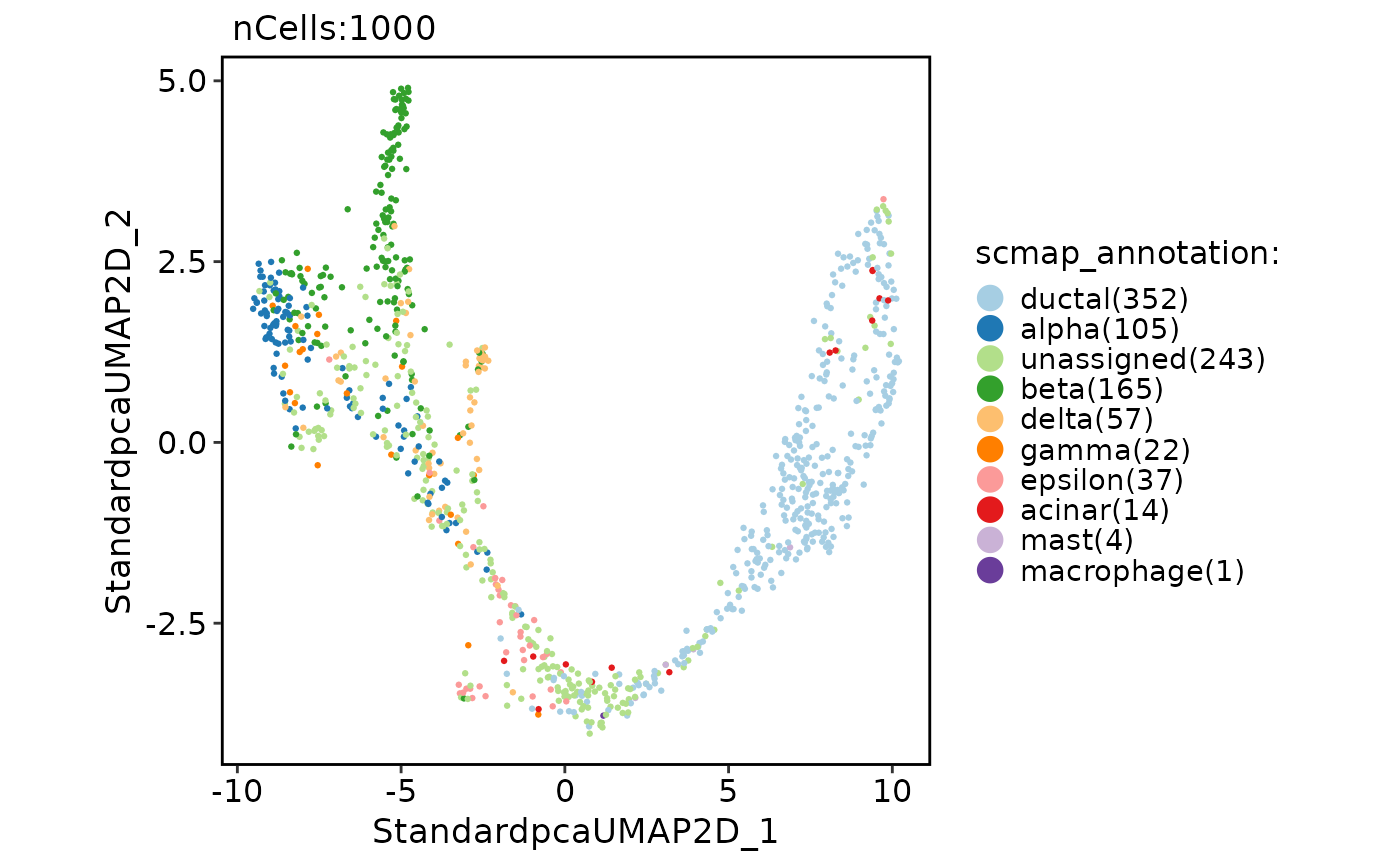
CellDimPlot(
pancreas_sub,
group.by = "scmap_annotation"
)
pancreas_sub <- RunScmap(
srt_query = pancreas_sub,
srt_ref = panc8_sub,
ref_group = "celltype",
method = "scmapCell"
)
#> ℹ [2026-07-02 09:51:51] Data type is log-normalized
#> ℹ [2026-07-02 09:51:51] Detected `srt_query` data type: "log_normalized_counts"
#> ℹ [2026-07-02 09:51:52] Data type is log-normalized
#> ℹ [2026-07-02 09:51:52] Detected `srt_ref` data type: "log_normalized_counts"
#> ℹ [2026-07-02 09:51:53] Perform selectFeatures
#> ℹ [2026-07-02 09:51:53] Perform indexCell
#> ℹ [2026-07-02 09:51:53] Perform scmapCell
#> ℹ [2026-07-02 09:51:54] Perform scmapCell2Cluster
CellDimPlot(
pancreas_sub,
group.by = "scmap_annotation"
)