Perform the enrichment analysis (GSEA) on the genes
Usage
RunGSEA(
srt = NULL,
group.by = NULL,
test.use = "wilcox",
DE_threshold = "p_val_adj < 0.05",
scoreType = "std",
geneID = NULL,
geneScore = NULL,
geneID_groups = NULL,
geneID_exclude = NULL,
IDtype = "symbol",
result_IDtype = "symbol",
species = "Homo_sapiens",
db = "GO_BP",
db_update = FALSE,
db_version = "latest",
db_combine = FALSE,
convert_species = TRUE,
Ensembl_version = NULL,
mirror = NULL,
features = NULL,
TERM2GENE = NULL,
TERM2NAME = NULL,
minGSSize = 10,
maxGSSize = 500,
unlimited_db = c("Chromosome", "GeneType", "TF", "Enzyme", "CSPA"),
GO_simplify = FALSE,
GO_simplify_cutoff = "p.adjust < 0.05",
simplify_method = "Wang",
simplify_similarityCutoff = 0.7,
cores = 1,
verbose = TRUE,
...
)Arguments
- srt
A
Seuratobject orSummarizedExperimentobject containing the results of differential expression analysis (RunDEtest()). If specified, the genes and groups will be extracted from the object automatically. If not specified, thegeneIDandgeneID_groupsarguments must be provided.- group.by
Name of one or more meta.data columns to group (color) cells by.
- test.use
A character vector specifying the test to be used in differential expression analysis. This argument is only used if
srtis specified.- DE_threshold
A character vector specifying the filter condition for differential expression analysis. This argument is only used if
srtis specified.- scoreType
This parameter defines the GSEA score type. Possible options are "std", "pos", "neg". By default ("std") the enrichment score is computed as in the original GSEA. The "pos" and "neg" score types are intended to be used for one-tailed tests (i.e. when one is interested only in positive ("pos") or negateive ("neg") enrichment).
- geneID
A character vector specifying the gene IDs.
- geneScore
A numeric vector that specifies the gene scores, for example, the log2(fold change) values of gene expression.
- geneID_groups
A factor vector specifying the group labels for each gene.
- geneID_exclude
A character vector specifying the gene IDs to be excluded from the analysis.
- IDtype
A character vector specifying the type of gene IDs in the
srtobject orgeneIDargument. This argument is used to convert the gene IDs to a different type ifIDtypeis different fromresult_IDtype.- result_IDtype
A character vector specifying the desired type of gene ID to be used in the output. This argument is used to convert the gene IDs from
IDtypetoresult_IDtype.- species
A character vector specifying the species for which the gene annotation databases should be prepared. Can be
"Homo_sapiens"or"Mus_musculus".- db
A character vector specifying the annotation sources to be included in the gene annotation databases. Can be one or more of
"GO", "GO_BP", "GO_CC", "GO_MF", "KEGG", "WikiPathway", "Reactome", "CORUM", "MP", "DO", "HPO", "PFAM", "CSPA", "Surfaceome", "SPRomeDB", "VerSeDa", "TFLink", "hTFtarget", "TRRUST", "JASPAR", "ENCODE", "MSigDB", "CellTalk", "CellChat", "Chromosome", "GeneType", "Enzyme", "TF", "CytoTRACE2". MSigDB subcollections can be requested as"MSigDB_<collection>", such as"MSigDB_H"for human Hallmark and"MSigDB_MH"for mouse Hallmark. Note:"CytoTRACE2"is species-independent and downloads pre-trained model data required by RunCytoTRACE.- db_update
Whether the gene annotation databases should be forcefully updated. If set to FALSE, the function will attempt to load the cached databases instead. Default is
FALSE.- db_version
A character vector specifying the version of the gene annotation databases to be retrieved. Default is
"latest".- db_combine
Whether to combine multiple databases into one. If
TRUE, all database specified bydbwill be combined as one named "Combined".- convert_species
Whether to use a species-converted database when the annotation is missing for the specified species. Default is
TRUE.- Ensembl_version
An integer specifying the Ensembl version. Default is
NULL. IfNULL, the latest version will be used.- mirror
Specify an Ensembl mirror to connect to. The valid options here are
"www","uswest","useast","asia".- features
A named list of feature lists for custom enrichment gene sets. If provided, it takes precedence over
TERM2GENEanddb.- TERM2GENE
A data frame specifying the gene-term mapping for a custom database. The first column should contain the term IDs, and the second column should contain the gene IDs.
- TERM2NAME
A data frame specifying the term-name mapping for a custom database. The first column should contain the term IDs, and the second column should contain the corresponding term names.
- minGSSize
The minimum size of a gene set to be considered in the enrichment analysis.
- maxGSSize
The maximum size of a gene set to be considered in the enrichment analysis.
- unlimited_db
A character vector specifying the names of databases that do not have size restrictions.
- GO_simplify
Whether to simplify the GO terms. If
TRUE, additional results with simplified GO terms will be returned.- GO_simplify_cutoff
A character vector specifying the filter condition for simplification of GO terms. This argument is only used if
GO_simplifyisTRUE.- simplify_method
A character vector specifying the method to be used for simplification of GO terms. This argument is only used if
GO_simplifyisTRUE.- simplify_similarityCutoff
The similarity cutoff for simplification of GO terms. This argument is only used if
GO_simplifyisTRUE.- cores
The number of cores to use for parallelization with foreach::foreach. Default is
1.- verbose
Whether to print the message. Default is
TRUE.- ...
Passed to other functions.
Value
If input is a Seurat object, returns the modified Seurat object with the enrichment result stored in the tools slot. If input is a geneID vector with or without geneID_groups, return the enrichment result directly. Enrichment result is a list with the following component:
enrichment: A data.frame containing all enrichment results.results: A list ofgseaResultobjects from the DOSE package.geneMap: A data.frame containing the ID mapping table for input gene IDs.input: A data.frame containing the input gene IDs and gene ID groups.DE_threshold: A specific threshold for differential expression analysis (only returned if input is a Seurat object).
Examples
term2gene <- data.frame(
Term = c(
rep("Endocrine markers", 5),
rep("Exocrine markers", 5),
rep("Ductal markers", 5)
),
symbol = c(
"INS", "GCG", "SST", "IAPP", "PCSK1",
"PRSS1", "CPA1", "CELA3A", "REG1A", "CTRB1",
"KRT19", "SOX9", "MUC1", "CFTR", "KRT7"
)
)
genes <- unique(term2gene$symbol)
scores <- seq_along(genes)
names(scores) <- genes
gsea_out <- RunGSEA(
geneID = genes,
geneScore = scores,
geneID_groups = rep("Cluster1", length(genes)),
TERM2GENE = term2gene,
minGSSize = 2
)
#> ℹ [2026-07-02 09:42:17] Start GSEA analysis
#> ! [2026-07-02 09:42:17] All values in the `geneScore` are greater than zero. Set scoreType = 'pos'
#> ℹ [2026-07-02 09:42:17] Preparing custom GSEA database from `TERM2GENE`/`TERM2NAME` ...
#> ℹ [2026-07-02 09:42:17] Prepared 15 ranked gene rows after ID mapping.
#> ℹ [2026-07-02 09:42:17] Running GSEA for 1 group/database combination(s) using 1 core(s) ...
#> ℹ [2026-07-02 09:42:17] Using 1 core
#> ℹ [2026-07-02 09:42:17] Running GSEA: group "Cluster1", database "custom", genes 15 ...
#> ℹ [2026-07-02 09:42:17] Finished GSEA: group "Cluster1", database "custom".
#> ℹ [2026-07-02 09:42:17] Building results
#> ✔ [2026-07-02 09:42:17] GSEA analysis done
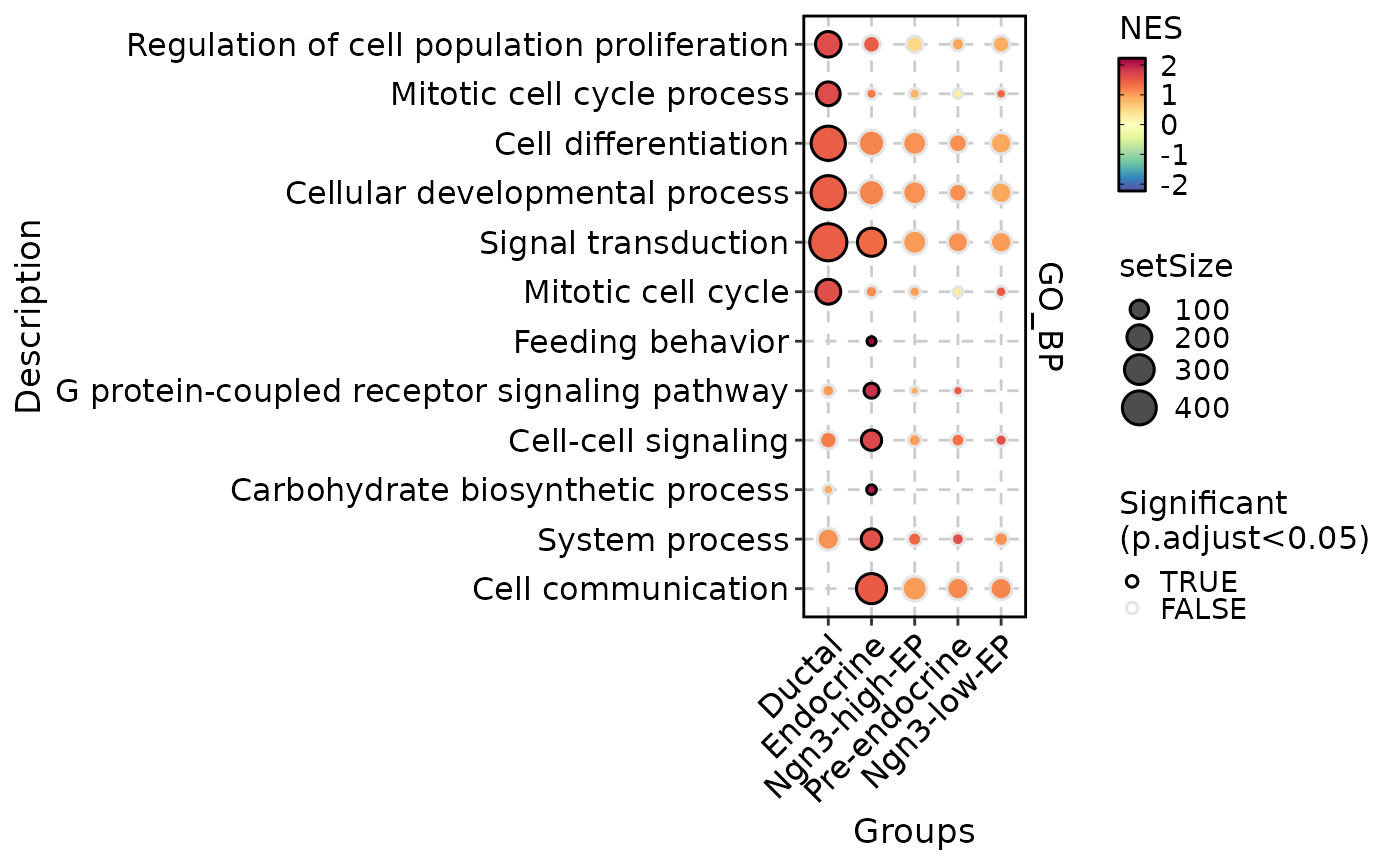
GSEAPlot(
res = gsea_out,
db = "custom",
plot_type = "comparison"
)
#> Warning: No shared levels found between `names(values)` of the manual scale and the
#> data's alpha values.