Run ambient RNA decontamination with decontX
Usage
RunDecontX(
srt,
assay = "RNA",
group.by = NULL,
batch = NULL,
background = NULL,
background_assay = NULL,
bg_batch = NULL,
assay_name = "decontXcounts",
store_assay = TRUE,
round_counts = FALSE,
data_type = NULL,
seed = 11,
...,
verbose = TRUE
)Arguments
- srt
A Seurat object.
- assay
The name of the assay to be used for decontamination. Default is
"RNA".- group.by
Cell cluster labels passed to
decontX::decontX(). Can beNULL, a meta.data column name, or a vector aligned to cells. Default isNULL.- batch
Batch labels passed to
decontX::decontX(). Can beNULL, a meta.data column name, or a vector aligned to cells. Default isNULL.- background
Optional background / empty-droplet input passed to
decontX::decontX(). Can be aSeuratobject,SingleCellExperiment, or count matrix. Default isNULL.- background_assay
Assay name used when
backgroundis aSeuratobject orSingleCellExperiment. Default isNULL, which falls back toassayforSeuratbackground and"counts"forSingleCellExperimentbackground.- bg_batch
Batch labels for
backgroundpassed todecontX::decontX(). Can beNULL, a metadata column name, or a vector aligned to the background droplets. Default isNULL.- assay_name
Name of the assay used to store decontaminated counts. Default is
"decontXcounts".- store_assay
Whether to store decontaminated counts as a new assay. Default is
TRUE.- round_counts
Whether to round decontaminated counts before creating the assay. Default is
FALSE.- data_type
Optional precomputed result from
CheckDataType()for the input assay. Primarily used internally to avoid repeated scans of the same count matrix across nested QC calls.- seed
Random seed for reproducibility. Default is
11.- ...
Additional arguments passed to
decontX::decontX().- verbose
Whether to print the message. Default is
TRUE.
Value
Returns a Seurat object with decontX contamination estimates stored in the meta.data, and optional decontaminated counts stored in a new assay.
Examples
data(pancreas_sub)
pancreas_sub <- standard_scop(pancreas_sub)
#> ℹ [2026-07-02 09:36:23] Start standard processing workflow...
#> ℹ [2026-07-02 09:36:24] Checking a list of <Seurat>...
#> ! [2026-07-02 09:36:24] Data 1/1 of the `srt_list` is "unknown"
#> ℹ [2026-07-02 09:36:24] Perform `NormalizeData()` with `normalization.method = 'LogNormalize'` on 1/1 of `srt_list`...
#> ℹ [2026-07-02 09:36:24] Perform `FindVariableFeatures()` on 1/1 of `srt_list`...
#> ℹ [2026-07-02 09:36:24] Use the separate HVF from `srt_list`
#> ℹ [2026-07-02 09:36:25] Number of available HVF: 2000
#> ℹ [2026-07-02 09:36:25] Finished check
#> ℹ [2026-07-02 09:36:25] Perform `ScaleData()`
#> ℹ [2026-07-02 09:36:25] Perform pca linear dimension reduction
#> ℹ [2026-07-02 09:36:25] Use stored estimated dimensions 1:23 for Standardpca
#> ℹ [2026-07-02 09:36:26] Perform `Seurat::FindClusters()` with `cluster_algorithm = 'louvain'` and `cluster_resolution = 0.6`
#> ℹ [2026-07-02 09:36:26] Reorder clusters...
#> ℹ [2026-07-02 09:36:26] Skip `log1p()` because `layer = data` is not "counts"
#> ℹ [2026-07-02 09:36:26] Perform umap nonlinear dimension reduction
#> ✔ [2026-07-02 09:36:32] Standard processing workflow completed
pancreas_sub <- RunDecontX(
pancreas_sub,
group.by = "CellType"
)
#> ℹ [2026-07-02 09:36:32] Running decontX
#> ℹ [2026-07-02 09:36:33] Data type is raw counts
#> Warning: 'librarySizeFactors' is deprecated.
#> Use 'scrapper::centerSizeFactors' instead.
#> See help("Deprecated")
#> Warning: 'normalizeCounts' is deprecated.
#> Use 'scrapper::normalizeCounts' instead.
#> See help("Deprecated")
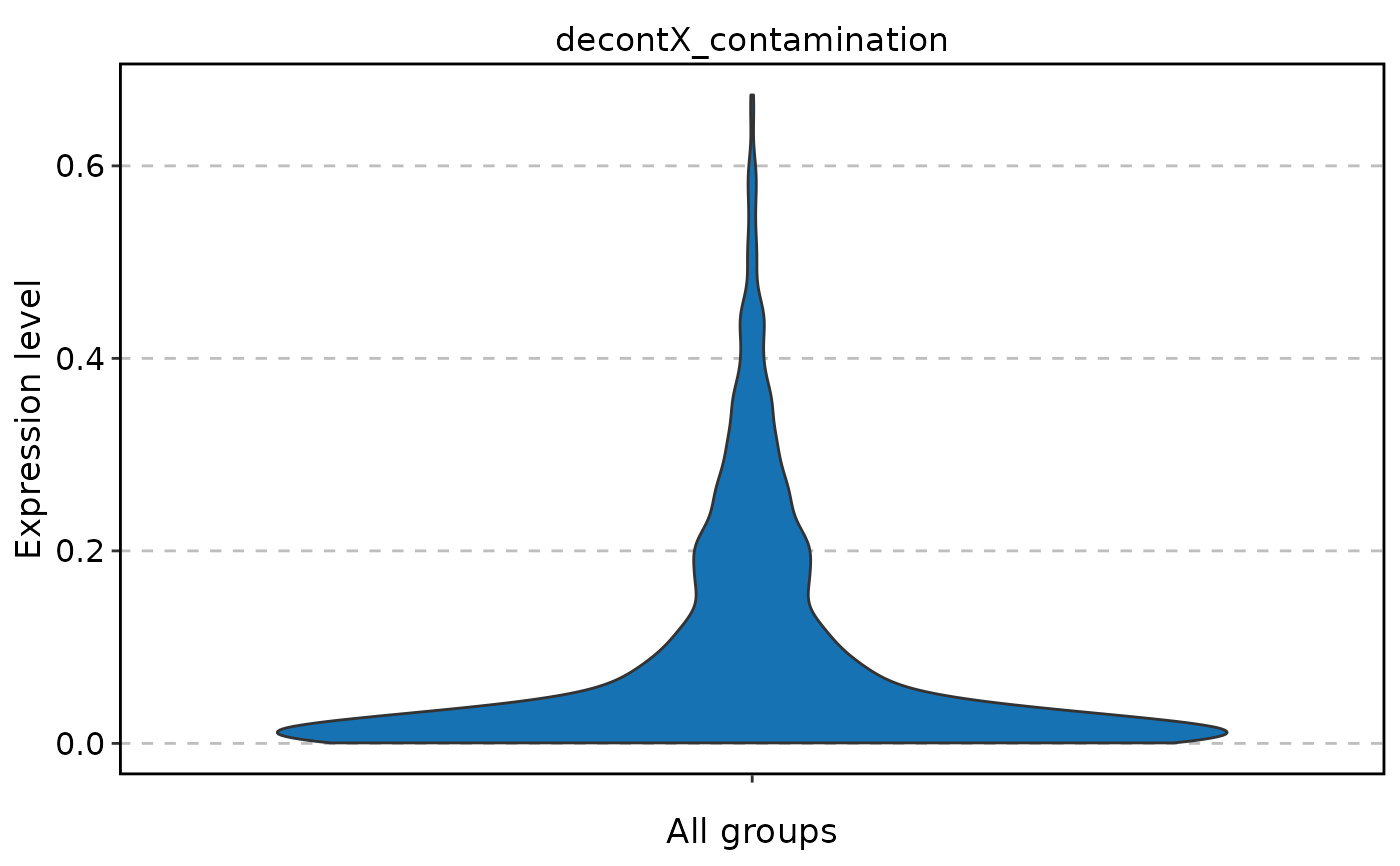
#> ℹ [2026-07-02 09:36:46] decontX contamination (median/mean/max): 0.0272 / 0.0875 / 0.6737
#> ℹ [2026-07-02 09:36:46] decontX assay stored as decontXcounts
#> ✔ [2026-07-02 09:36:46] decontX decontamination completed
FeatureStatPlot(
pancreas_sub,
stat.by = "decontX_contamination"
)
#> Warning: No shared levels found between `names(values)` of the manual scale and the
#> data's colour values.
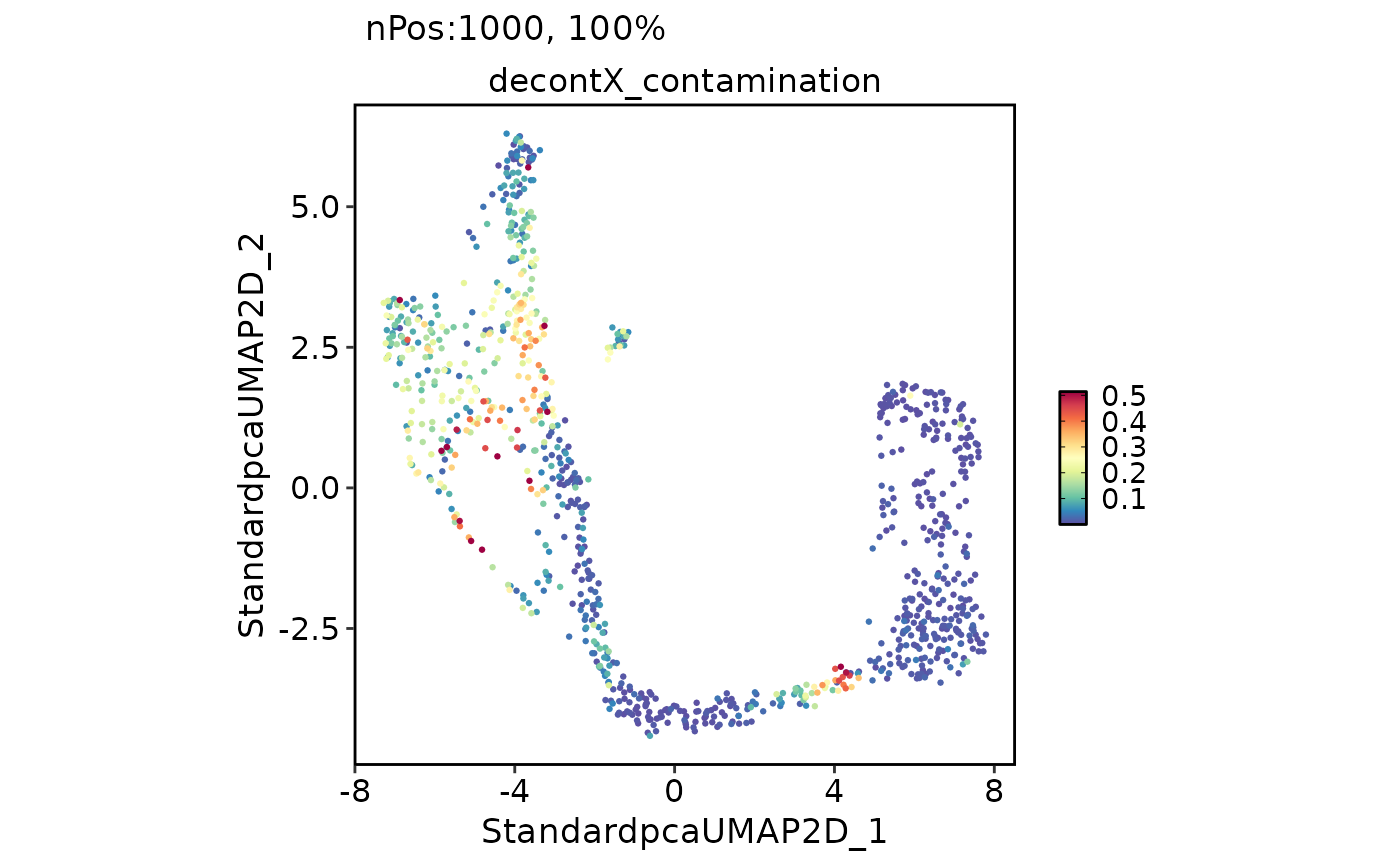
 FeatureDimPlot(
pancreas_sub,
features = "decontX_contamination"
)
FeatureDimPlot(
pancreas_sub,
features = "decontX_contamination"
)