Calculate common spot-level QC metrics for spatial transcriptomics data and label failed spots without running single-cell-specific checks such as doublet calling or ambient RNA decontamination.
Usage
RunSpotQC(
srt,
assay = NULL,
return_filtered = FALSE,
qc_metrics = c("outlier", "umi", "gene", "mito"),
outlier_threshold = c("log10_nCount:lower:3", "log10_nFeature:lower:3",
"spot_featurecount_dist:lower:3"),
outlier_n = 1,
UMI_threshold = 500,
gene_threshold = 200,
mito_threshold = 20,
mito_pattern = c("MT-", "Mt-", "mt-"),
mito_gene = NULL,
verbose = TRUE,
seed = 11
)Arguments
- srt
A Seurat object.
- assay
Which assay to use. If
NULL, the default assay of the Seurat object will be used. When the object also containsChromatinAssay, the default assay and additionalChromatinAssaywill be preprocessed sequentially.- return_filtered
Logical indicating whether to return a spot-filtered Seurat object. Default is
FALSE.- qc_metrics
QC metrics to apply. Available metrics are
"outlier","umi","gene", and"mito".- outlier_threshold
Character vector specifying outlier thresholds as
"metric:direction:nmads". Available default metrics are"log10_nCount","log10_nFeature", and"spot_featurecount_dist".- outlier_n
Minimum number of outlier metrics required to fail a spot.
- UMI_threshold
Minimum UMI count required to pass
"umi"QC.- gene_threshold
Minimum detected gene count required to pass
"gene"QC.- mito_threshold
Maximum mitochondrial percentage allowed by
"mito"QC.- mito_pattern
Regex patterns used to identify mitochondrial genes.
- mito_gene
Optional explicit mitochondrial gene vector. When provided,
mito_patternis ignored.- verbose
Whether to print the message. Default is
TRUE.- seed
Random seed for reproducibility.
Examples
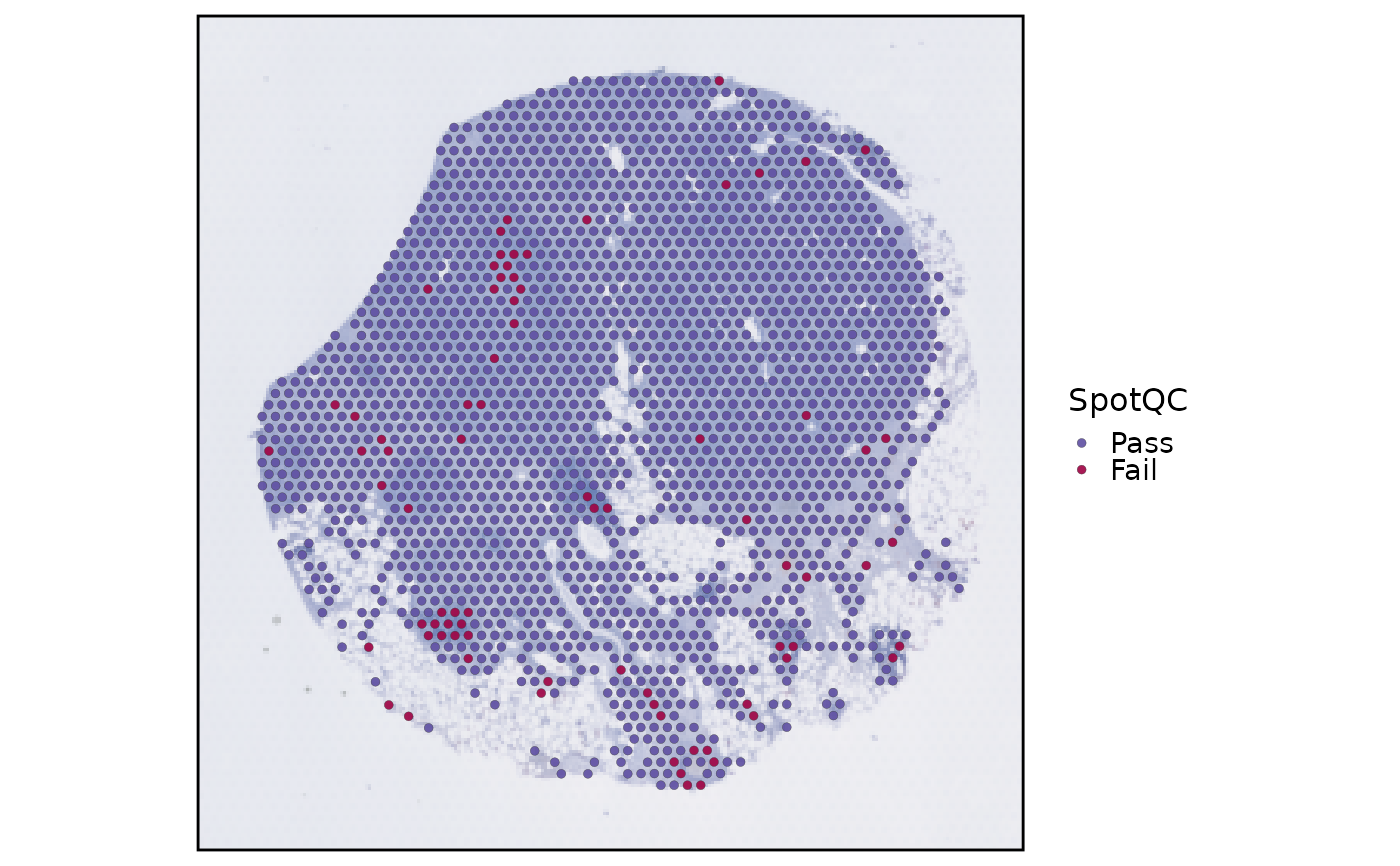
data(visium_human_pancreas_sub)
spatial <- RunSpotQC(
visium_human_pancreas_sub,
assay = "Spatial"
)
#> ◌ [2026-07-02 10:09:07] Running spot-level quality control
#> ✔ [2026-07-02 10:09:08] 1907 spots passed QC and 79 spots failed QC
SpatialSpotPlot(spatial, group.by = "SpotQC")