Run MDS (multi-dimensional scaling)
Usage
RunMDS(object, ...)
# S3 method for class 'Seurat'
RunMDS(
object,
assay = NULL,
layer = "data",
features = NULL,
nmds = 50,
dist.method = "euclidean",
mds.method = "cmdscale",
rev.mds = FALSE,
reduction.name = "mds",
reduction.key = "MDS_",
verbose = TRUE,
seed.use = 11,
...
)
# S3 method for class 'Assay'
RunMDS(
object,
assay = NULL,
layer = "data",
features = NULL,
nmds = 50,
dist.method = "euclidean",
mds.method = "cmdscale",
rev.mds = FALSE,
reduction.key = "MDS_",
verbose = TRUE,
seed.use = 11,
...
)
# S3 method for class 'Assay5'
RunMDS(
object,
assay = NULL,
layer = "data",
features = NULL,
nmds = 50,
dist.method = "euclidean",
mds.method = "cmdscale",
rev.mds = FALSE,
reduction.key = "MDS_",
verbose = TRUE,
seed.use = 11,
...
)
# Default S3 method
RunMDS(
object,
assay = NULL,
layer = "data",
nmds = 50,
dist.method = "euclidean",
mds.method = "cmdscale",
rev.mds = FALSE,
reduction.key = "MDS_",
verbose = TRUE,
seed.use = 11,
...
)Arguments
- object
An object. This can be a Seurat object, an assay object, or a matrix-like object.
- ...
Additional arguments to be passed to stats::cmdscale, MASS::isoMDS or MASS::sammon.
- assay
Which assay to use. If
NULL, the default assay of the Seurat object will be used. When the object also containsChromatinAssay, the default assay and additionalChromatinAssaywill be preprocessed sequentially.- layer
Which layer to use. Default is
data.- features
A character vector of features to use. Default is
NULL.- nmds
The number of dimensions to be computed. Default is
50.- dist.method
The distance metric to be used. Currently supported values are
"euclidean","chisquared","kullback","jeffreys","jensen","manhattan","maximum","canberra","minkowski", and"hamming". Default is"euclidean".- mds.method
The MDS algorithm to be used. Currently supported values are
"cmdscale","isoMDS", and"sammon". Default is"cmdscale".- rev.mds
Whether to perform reverse MDS (i.e., transpose the input matrix) before running the analysis. Default is
FALSE.- reduction.name
The name of the reduction to be stored in the Seurat object. Default is
"mds".- reduction.key
The prefix for the column names of the basis vectors. Default is
"MDS_".- verbose
Whether to print the message. Default is
TRUE.- seed.use
Random seed for reproducibility. Default is
11.
Examples
data(pancreas_sub)
pancreas_sub <- standard_scop(pancreas_sub)
#> ℹ [2026-07-02 09:45:38] Start standard processing workflow...
#> ℹ [2026-07-02 09:45:39] Checking a list of <Seurat>...
#> ! [2026-07-02 09:45:39] Data 1/1 of the `srt_list` is "unknown"
#> ℹ [2026-07-02 09:45:39] Perform `NormalizeData()` with `normalization.method = 'LogNormalize'` on 1/1 of `srt_list`...
#> ℹ [2026-07-02 09:45:39] Perform `FindVariableFeatures()` on 1/1 of `srt_list`...
#> ℹ [2026-07-02 09:45:39] Use the separate HVF from `srt_list`
#> ℹ [2026-07-02 09:45:39] Number of available HVF: 2000
#> ℹ [2026-07-02 09:45:39] Finished check
#> ℹ [2026-07-02 09:45:39] Perform `ScaleData()`
#> ℹ [2026-07-02 09:45:39] Perform pca linear dimension reduction
#> ℹ [2026-07-02 09:45:40] Use stored estimated dimensions 1:23 for Standardpca
#> ℹ [2026-07-02 09:45:40] Perform `Seurat::FindClusters()` with `cluster_algorithm = 'louvain'` and `cluster_resolution = 0.6`
#> ℹ [2026-07-02 09:45:40] Reorder clusters...
#> ℹ [2026-07-02 09:45:40] Skip `log1p()` because `layer = data` is not "counts"
#> ℹ [2026-07-02 09:45:40] Perform umap nonlinear dimension reduction
#> ✔ [2026-07-02 09:45:47] Standard processing workflow completed
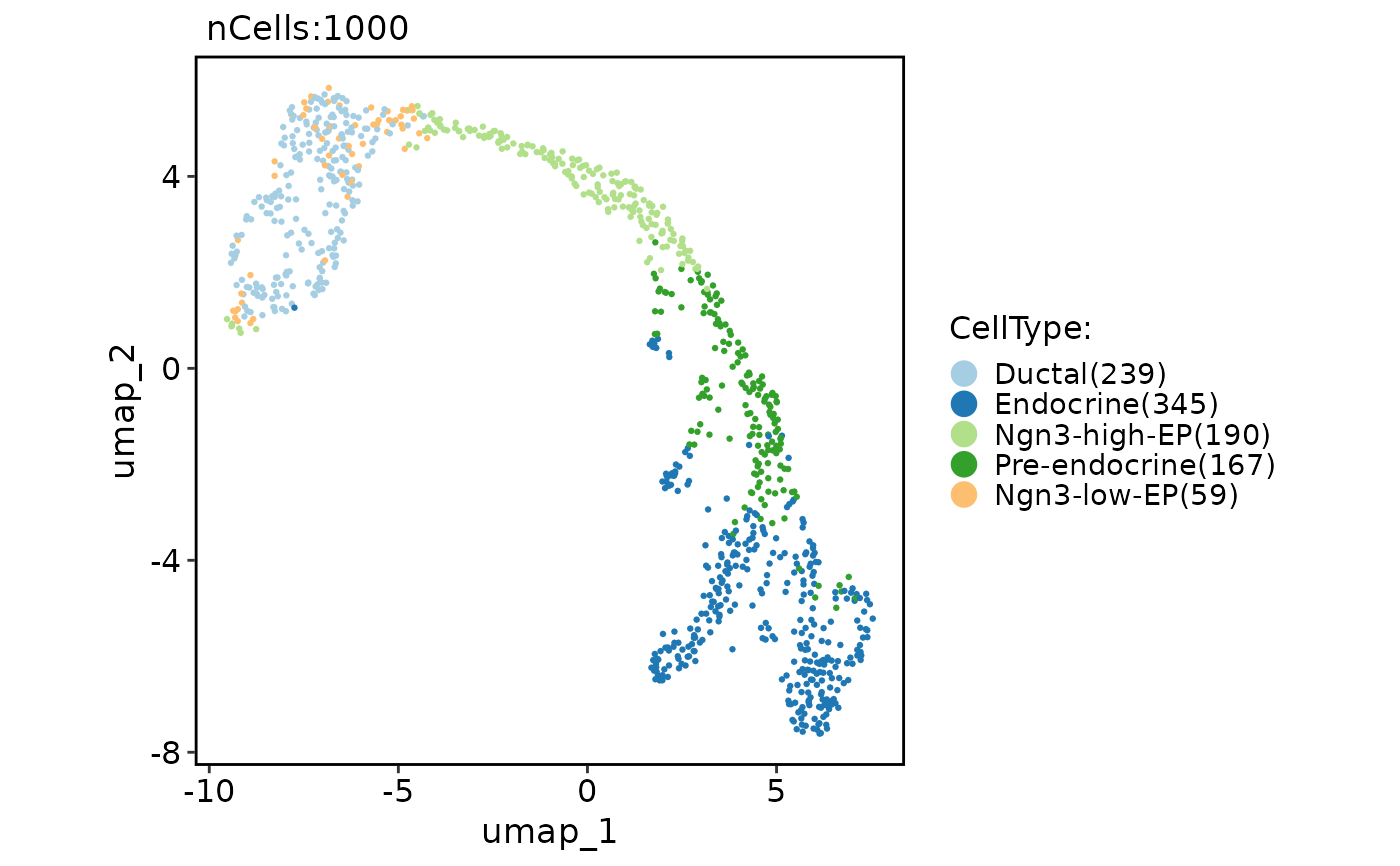
pancreas_sub <- RunMDS(pancreas_sub)
CellDimPlot(
pancreas_sub,
group.by = "CellType",
reduction = "mds"
)