Run CytoSPACE spatial assignment
Usage
RunCytoSPACE(
srt,
reference,
reference_label,
assay = NULL,
reference_assay = NULL,
layer = "counts",
reference_layer = "counts",
features = NULL,
cell_fractions = NULL,
n_cells_per_spot = NULL,
mean_cell_numbers = 5,
scRNA_max_transcripts_per_cell = 1500,
sampling_method = "duplicates",
seed = 1,
prefix = "CytoSPACE",
store_results = TRUE,
verbose = TRUE
)Arguments
- srt
A Seurat object.
- reference
Reference
Seuratobject containing annotated single cells.- reference_label
Metadata column in
referencewith cell type labels.- assay
Which assay to use. If
NULL, the default assay of the Seurat object will be used. When the object also containsChromatinAssay, the default assay and additionalChromatinAssaywill be preprocessed sequentially.- reference_assay
Assay used in
reference.- layer, reference_layer
Assay layers used for spatial and reference expression.
- features
Features used for assignment. If
NULL, shared features are used.- cell_fractions
Optional cell-type fractions. Provide a named numeric vector, one-row matrix/data.frame, or a spot-by-cell-type matrix/data.frame. Spot-level rows are aggregated to the global composition used by the default CytoSPACE assignment workflow.
- n_cells_per_spot
Optional number of cells assigned to each spatial spot. If
NULL, counts are estimated from spatial RNA reads withmean_cell_numbers.- mean_cell_numbers
Mean number of cells per spot. Default
5, matching the CytoSPACE Visium default.- scRNA_max_transcripts_per_cell
Maximum reference transcripts per cell before assignment. Default
1500, matching CytoSPACE.- sampling_method
Sampling method. Only
"duplicates"is supported in the package runtime.- seed
Random seed used for deterministic reference downsampling and duplicate sampling.
- prefix
Prefix for metadata columns.
- store_results
Whether to store detailed assignment results in
srt@tools.- verbose
Whether to print the message. Default is
TRUE.
Value
A Seurat object with CytoSPACE metadata columns and detailed
results stored in srt@tools[["CytoSPACE"]].
Examples
data(visium_human_pancreas_sub)
data(pancreas_sub)
features_use <- intersect(
rownames(visium_human_pancreas_sub),
rownames(pancreas_sub)
)
spatial <- RunCytoSPACE(
visium_human_pancreas_sub,
reference = pancreas_sub,
reference_label = "CellType",
features = features_use,
mean_cell_numbers = 1
)
#> ℹ [2026-07-02 09:31:33] Estimated CytoSPACE cell type fractions for 5 reference labels
#> ℹ [2026-07-02 09:31:33] Run CytoSPACE with 4 features, 686 sampled reference cells, and 1986 spatial spots
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:33] Assign sampled reference cells to spatial spots ■■■■■■…
#> ℹ [2026-07-02 09:31:41] Build CytoSPACE assignment summaries
#> ℹ [2026-07-02 09:31:41] CytoSPACE assignments stored in `srt@tools[['CytoSPACE']]` and metadata prefix "CytoSPACE"
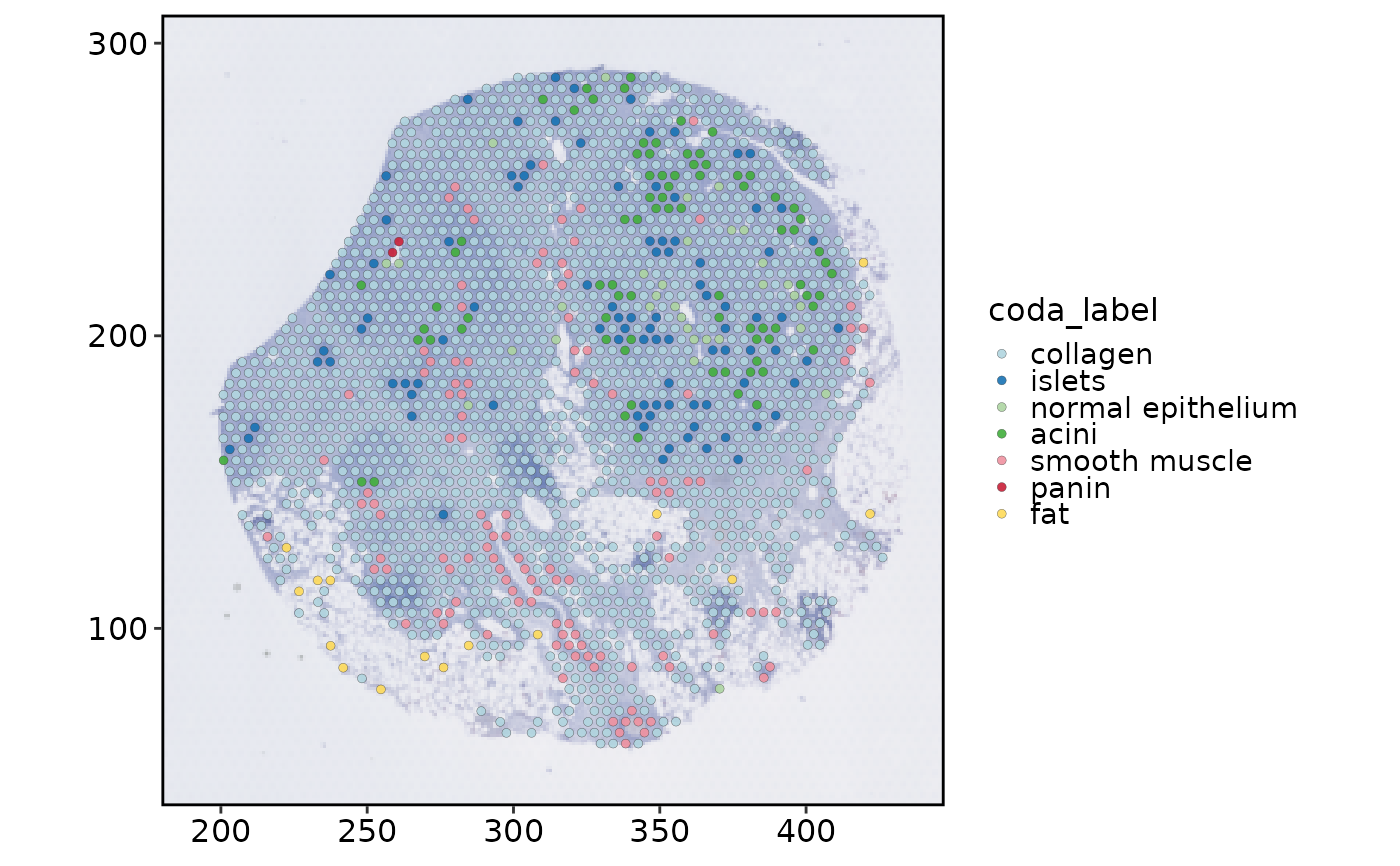
SpatialSpotPlot(
visium_human_pancreas_sub,
group.by = "coda_label",
theme_use = "theme_scop"
)
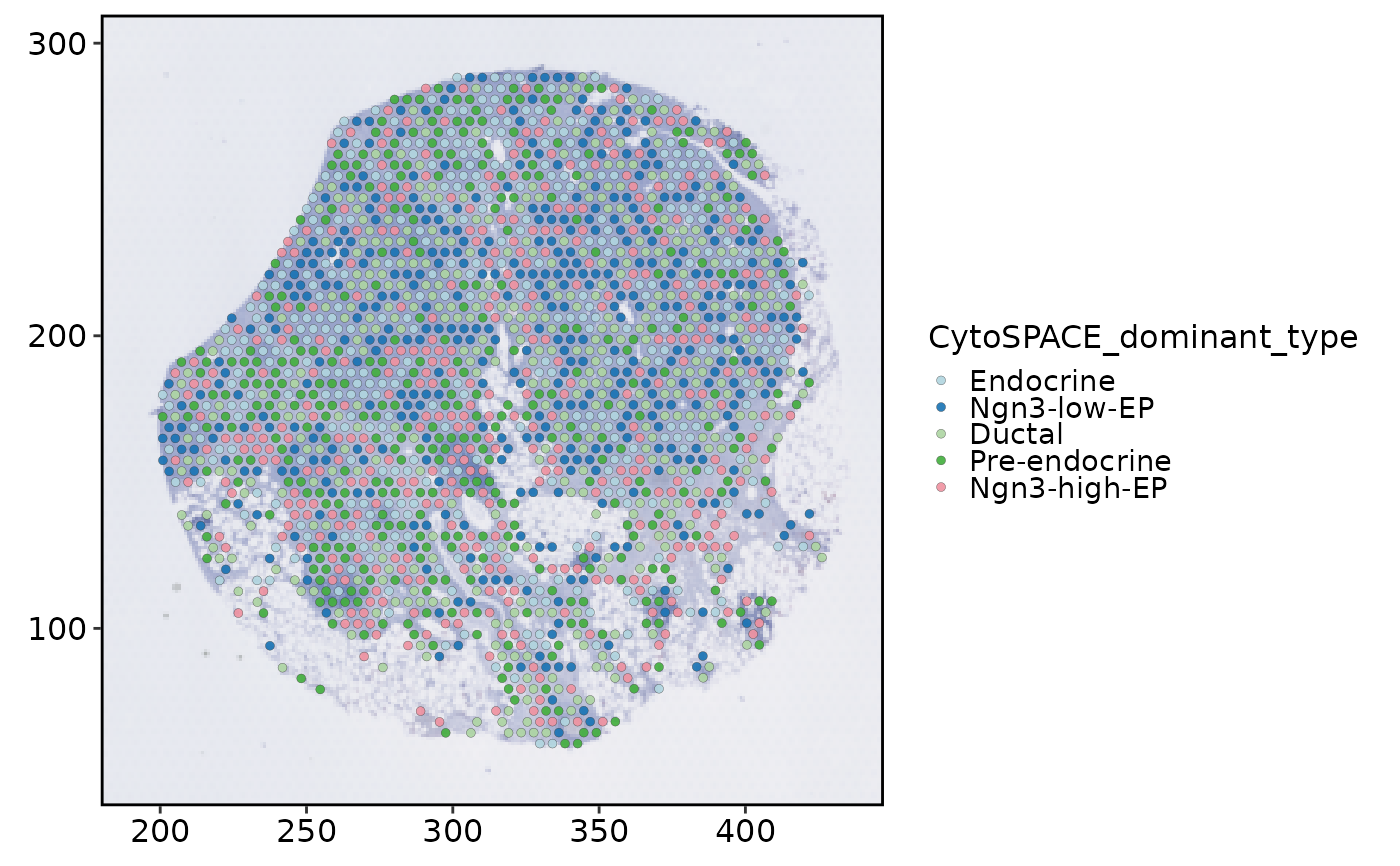
 SpatialSpotPlot(
spatial,
group.by = "CytoSPACE_dominant_type",
theme_use = "theme_scop"
)
SpatialSpotPlot(
spatial,
group.by = "CytoSPACE_dominant_type",
theme_use = "theme_scop"
)