Run cell-level quality control
Usage
RunCellQC(
srt,
assay = "RNA",
split.by = NULL,
group.by = NULL,
return_filtered = FALSE,
qc_metrics = c("doublets", "decontX", "atac", "outlier", "umi", "gene", "mito", "ribo",
"ribo_mito_ratio", "species"),
db_method = "scDblFinder",
db_rate = NULL,
db_coefficient = 0.01,
decontX_threshold = NULL,
decontX_batch = NULL,
decontX_background = NULL,
decontX_background_assay = NULL,
decontX_bg_batch = NULL,
decontX_assay_name = "decontXcounts",
decontX_store_assay = FALSE,
decontX_round_counts = TRUE,
decontX_args = list(),
atac_args = list(),
outlier_threshold = c("log10_nCount:lower:2.5", "log10_nCount:higher:5",
"log10_nFeature:lower:2.5", "log10_nFeature:higher:5", "featurecount_dist:lower:2.5"),
outlier_n = 1,
UMI_threshold = 3000,
gene_threshold = 1000,
mito_threshold = 20,
mito_pattern = c("MT-", "Mt-", "mt-"),
mito_gene = NULL,
ribo_threshold = 50,
ribo_pattern = c("RP[SL]\\d+\\w{0,1}\\d*$", "Rp[sl]\\d+\\w{0,1}\\d*$",
"rp[sl]\\d+\\w{0,1}\\d*$"),
ribo_gene = NULL,
ribo_mito_ratio_range = c(1, Inf),
species = NULL,
species_gene_prefix = NULL,
species_percent = 95,
seed = 11,
verbose = TRUE,
hb_range = c(0, 5),
hb_pattern = c("HB[^P]", "Hb[^p]", "hb[^p]"),
hb_gene = NULL,
qc_features = list()
)Arguments
- srt
A Seurat object.
- assay
The name of the assay to be used for doublet-calling. Default is
"RNA".- split.by
Name of a meta.data column used to split the object before QC. Default is
NULL. When specified, QC and doublet-calling are performed separately within each split object and merged back afterward.- group.by
Group labels passed to
RunDecontX()when"decontX"is included inqc_metrics. Can beNULL, a meta.data column name, or a vector aligned to cells. Default isNULL.- return_filtered
Logical indicating whether to return a cell-filtered Seurat object. Default is
FALSE.- qc_metrics
A character vector specifying the quality control metrics to be applied. Available metrics are
"doublets","decontX","atac","outlier","umi","gene","mito","ribo","hb","ribo_mito_ratio","species", and any rule name supplied inqc_features. Default isc("doublets", "decontX", "outlier", "umi", "gene", "mito", "ribo", "ribo_mito_ratio", "species"). ForChromatinAssay, if.arg qc_metricsis not supplied, the default is"atac".- db_method
Method used for doublet-calling. Can be one of
"scDblFinder","Scrublet","DoubletDetection","scds_cxds","scds_bcds","scds_hybrid". The resulting doublet labels are aggregated afterward intodb_qcand do not affect the thresholds used by the other QC metrics.- db_rate
The expected doublet rate. Default is calculated as
ncol(srt) / 1000 * 0.01.- db_coefficient
The coefficient used to calculate the doublet rate. Default is
0.01. Doublet rate is calculated asncol(srt) / 1000 * db_coefficient.- decontX_threshold
Optional contamination threshold used to filter cells after running
RunDecontX(). Cells withdecontX_contaminationgreater than this value are marked as failed indecontX_qc. Default isNULL, which computes decontX results without filtering cells by contamination.- decontX_batch
Batch labels passed to
RunDecontX()when"decontX"is included inqc_metrics. Default isNULL.- decontX_background
Optional background / empty-droplet input passed to
RunDecontX()when"decontX"is included inqc_metrics. Default isNULL.- decontX_background_assay
Assay name used when
decontX_backgroundis aSeuratobject orSingleCellExperiment. Default isNULL.- decontX_bg_batch
Batch labels for
decontX_backgroundpassed toRunDecontX(). Default isNULL.- decontX_assay_name
Name of the assay used to store decontaminated counts from
RunDecontX(). Default is"decontXcounts".- decontX_store_assay
Whether to store decontaminated counts as a new assay when running
RunDecontX(). Default isFALSE.- decontX_round_counts
Whether to round decontaminated counts before creating the assay in
RunDecontX(). Default isTRUE.- decontX_args
A named list of additional advanced arguments passed to
RunDecontX()when"decontX"is included inqc_metrics. ExplicitdecontX_*parameters are preferred for common options and take precedence when both are supplied. Default islist().- atac_args
A named list of additional arguments passed to
RunATACQC()when"atac"is included inqc_metrics. Threshold arguments fromRunATACQC()are used to label failed cells inatac_qc, but filtering is deferred toRunCellQC(). Default islist().- outlier_threshold
A character vector specifying the outlier threshold. Default is
c("log10_nCount:lower:2.5", "log10_nCount:higher:5", "log10_nFeature:lower:2.5", "log10_nFeature:higher:5", "featurecount_dist:lower:2.5").- outlier_n
Minimum number of outlier metrics that meet the conditions for determining outlier cells. Default is
1.- UMI_threshold
UMI number threshold. Cells that exceed this threshold will be considered as kept. Default is
3000.- gene_threshold
Gene number threshold. Cells that exceed this threshold will be considered as kept. Default is
1000.- mito_threshold
Percentage of UMI counts of mitochondrial genes. Cells that exceed this threshold will be considered as discarded. Default is
20.- mito_pattern
Regex patterns to match the mitochondrial genes. Default is
c("MT-", "Mt-", "mt-").- mito_gene
A defined mitochondrial genes. If features provided, will ignore the
mito_patternmatching. Default isNULL.- ribo_threshold
Percentage of UMI counts of ribosomal genes. Cells that exceed this threshold will be considered as discarded. Default is
50.- ribo_pattern
Regex patterns to match the ribosomal genes. Default is
c("RP[SL]\\d+\\w{0,1}\\d*$", "Rp[sl]\\d+\\w{0,1}\\d*$", "rp[sl]\\d+\\w{0,1}\\d*$").- ribo_gene
A defined ribosomal genes. If features provided, will ignore the
ribo_patternmatching. Default isNULL.- ribo_mito_ratio_range
A numeric vector specifying the range of ribosomal/mitochondrial gene expression ratios for ribo_mito_ratio outlier cells. Default is
c(1, Inf).- species
Species used as the suffix of the QC metrics. The first is the species of interest. Default is
NULL.- species_gene_prefix
Species gene prefix used to calculate QC metrics for each species. Default is
NULL.- species_percent
Percentage of UMI counts of the first species. Cells that exceed this threshold will be considered as kept. Default is
95.- seed
Random seed for reproducibility. Default is
11.- verbose
Whether to print the message. Default is
TRUE.- hb_range
A numeric vector specifying the accepted percentage range for hemoglobin features. Values outside the range fail
hb_qcwhen"hb"is included inqc_metrics. Default isc(0, 5).- hb_pattern
Regex patterns used to match hemoglobin features. The defaults match common human, mouse, and lower-case symbols while excluding
HBP/hbpgenes. Default isc("HB[^P]", "Hb[^p]", "hb[^p]").- hb_gene
A defined set of hemoglobin features. When supplied, these features are used instead of
hb_pattern. Default isNULL.- qc_features
A named list defining additional feature-percentage QC rules. Each rule must contain exactly one of
features(a character vector) orpattern(one or more regex patterns), plus a numericrangeof length two. Providing a rule computespercent.<name>; the rule filters cells and creates<name>_qconly when<name>is included inqc_metrics. Patterns are matched directly against assay feature names and do not receive species prefixes. Rule names cannot collide with built-in QC columns. Default islist().
Examples
data(pancreas_sub)
pancreas_sub <- standard_scop(pancreas_sub)
#> ℹ [2026-07-20 18:34:33] Start standard processing workflow...
#> ℹ [2026-07-20 18:34:33] Checking a list of <Seurat>...
#> ! [2026-07-20 18:34:33] Data 1/1 of the `srt_list` is "unknown"
#> ℹ [2026-07-20 18:34:33] Perform `NormalizeData()` with `normalization.method = 'LogNormalize'` on 1/1 of `srt_list`...
#> ℹ [2026-07-20 18:34:33] Perform `FindVariableFeatures()` on 1/1 of `srt_list`...
#> ℹ [2026-07-20 18:34:33] Use the separate HVF from `srt_list`
#> ℹ [2026-07-20 18:34:33] Number of available HVF: 2000
#> ℹ [2026-07-20 18:34:33] Finished check
#> ℹ [2026-07-20 18:34:33] Perform `ScaleData()`
#> ℹ [2026-07-20 18:34:34] Perform pca linear dimension reduction
#> ℹ [2026-07-20 18:34:35] Use stored estimated dimensions 1:23 for Standardpca
#> ℹ [2026-07-20 18:34:36] Perform `Seurat::FindClusters()` with `cluster_algorithm = 'louvain'` and `cluster_resolution = 0.6`
#> ℹ [2026-07-20 18:34:36] Reorder clusters...
#> ℹ [2026-07-20 18:34:36] Skip `log1p()` because `layer = data` is not "counts"
#> ℹ [2026-07-20 18:34:36] Perform umap nonlinear dimension reduction
#> ✔ [2026-07-20 18:34:40] Standard processing workflow completed
pancreas_sub <- RunCellQC(
pancreas_sub,
qc_metrics = c("umi", "gene", "example_set"),
qc_features = list(
example_set = list(
features = head(rownames(pancreas_sub), 5),
range = c(0, 0)
)
)
)
#> ◌ [2026-07-20 18:34:40] Running cell-level quality control
#> ℹ [2026-07-20 18:34:40] Data type is raw counts
#> ✔ [2026-07-20 18:34:41] ● Total cells: 1000
#> ✔ ◉ 849 cells remained
#> ✔ ◯ 151 cells filtered out:
#> ✔ ◯ 0 low-UMI cells
#> ✔ ◯ 0 low-gene cells
#> ✔ ◯ 151 example_set feature-QC outlier cells
head(pancreas_sub@meta.data[, c("percent.hb", "percent.example_set")])
#> percent.hb percent.example_set
#> AAACCTGAGCCTTGAT 0.00000000 0.01414227
#> AAACCTGGTAAGTGGC 0.00000000 0.00000000
#> AAACGGGAGATATGGT 0.01614466 0.00000000
#> AAACGGGCAAAGAATC 0.00000000 0.00000000
#> AAACGGGGTACAGTTC 0.01089087 0.00000000
#> AAACGGGTCAGCTCTC 0.00000000 0.00000000
# Hemoglobin expression can be biological signal in erythroid samples;
# include "hb" in qc_metrics only when HB-based filtering is appropriate.
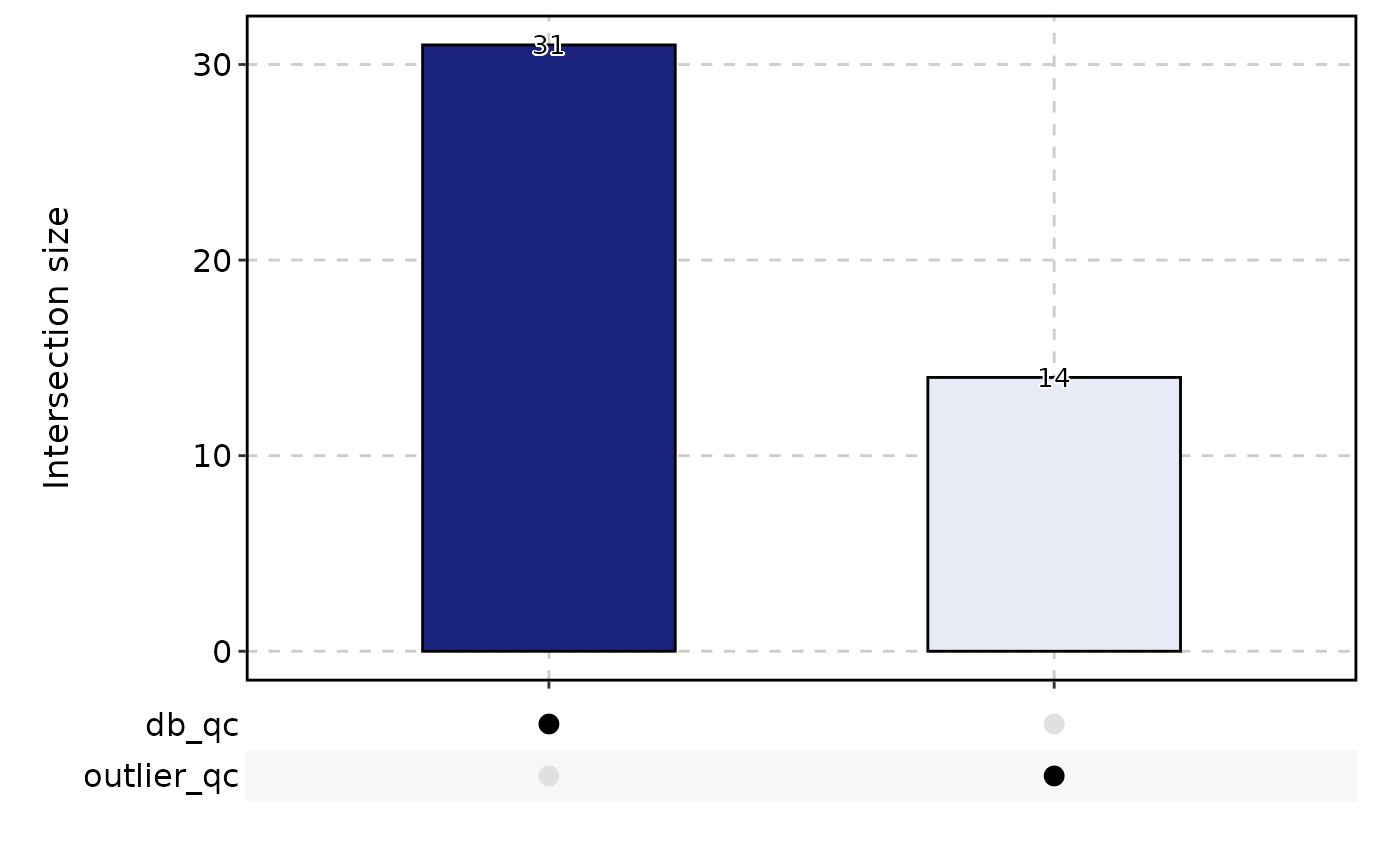
CellStatPlot(
pancreas_sub,
stat.by = c(
"umi_qc", "gene_qc", "example_set_qc"
),
plot_type = "upset",
stat_level = "Fail"
)
#> ! [2026-07-20 18:34:41] `stat_type` is forcibly set to "count" when plot "sankey", "chord", "venn", and "upset"
#> Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
#> ℹ Please use `linewidth` instead.
#> ℹ The deprecated feature was likely used in the ggupset package.
#> Please report the issue at <https://github.com/const-ae/ggupset/issues>.
#> `geom_line()`: Each group consists of only one observation.
#> ℹ Do you need to adjust the group aesthetic?
#> `geom_line()`: Each group consists of only one observation.
#> ℹ Do you need to adjust the group aesthetic?