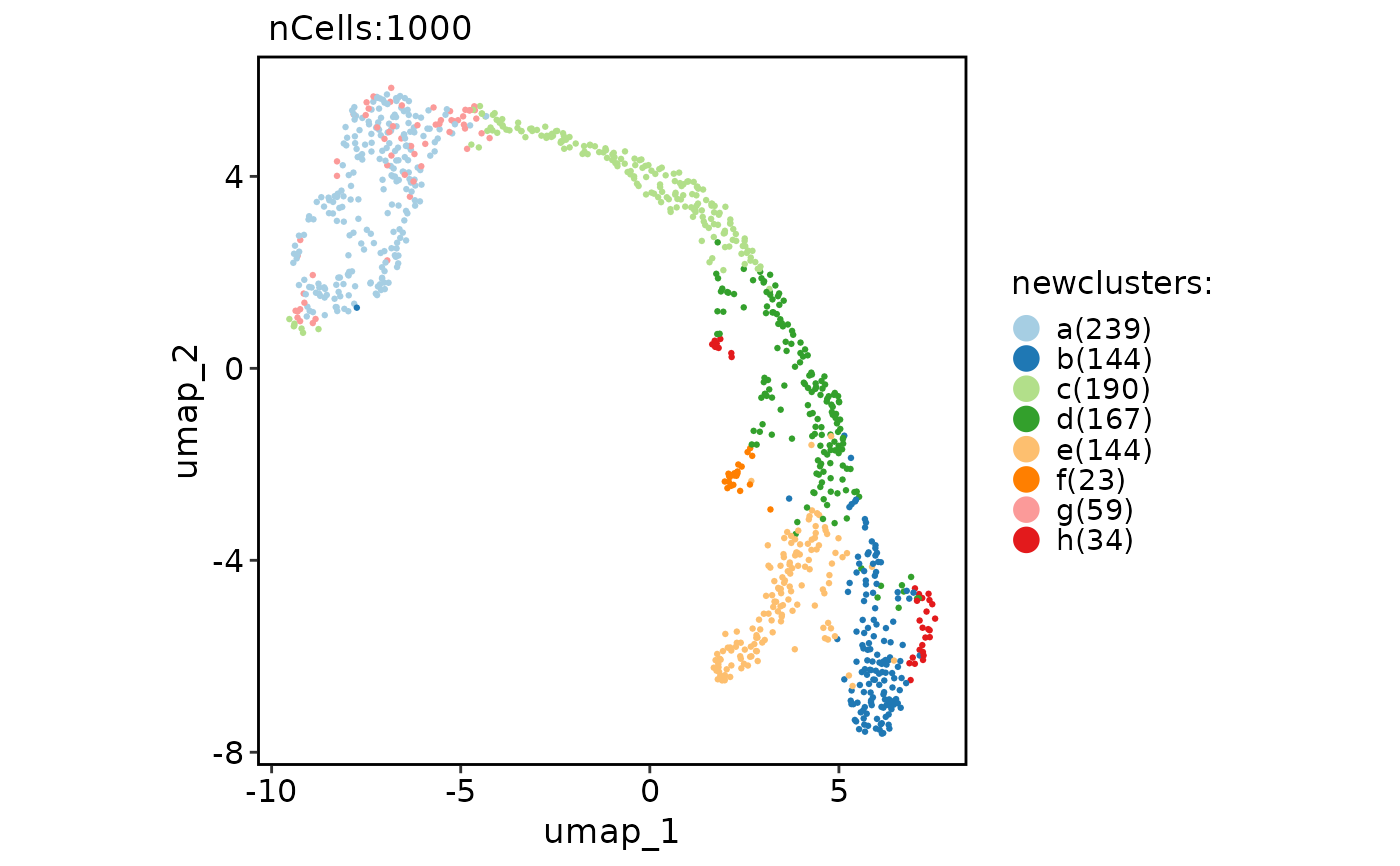
Rename clusters for the Seurat object
Usage
RenameClusters(
srt,
group.by,
nameslist = list(),
name = "newclusters",
keep_levels = FALSE,
verbose = TRUE
)Examples
data(pancreas_sub)
# Rename all clusters
pancreas_sub <- standard_scop(pancreas_sub)
#> ℹ [2026-07-02 09:20:04] Start standard processing workflow...
#> ℹ [2026-07-02 09:20:05] Checking a list of <Seurat>...
#> ! [2026-07-02 09:20:05] Data 1/1 of the `srt_list` is "unknown"
#> ℹ [2026-07-02 09:20:05] Perform `NormalizeData()` with `normalization.method = 'LogNormalize'` on 1/1 of `srt_list`...
#> ℹ [2026-07-02 09:20:05] Perform `FindVariableFeatures()` on 1/1 of `srt_list`...
#> ℹ [2026-07-02 09:20:06] Use the separate HVF from `srt_list`
#> ℹ [2026-07-02 09:20:06] Number of available HVF: 2000
#> ℹ [2026-07-02 09:20:06] Finished check
#> ℹ [2026-07-02 09:20:06] Perform `ScaleData()`
#> ℹ [2026-07-02 09:20:06] Perform pca linear dimension reduction
#> ℹ [2026-07-02 09:20:07] Use stored estimated dimensions 1:23 for Standardpca
#> ℹ [2026-07-02 09:20:07] Perform `Seurat::FindClusters()` with `cluster_algorithm = 'louvain'` and `cluster_resolution = 0.6`
#> ℹ [2026-07-02 09:20:07] Reorder clusters...
#> ℹ [2026-07-02 09:20:07] Skip `log1p()` because `layer = data` is not "counts"
#> ℹ [2026-07-02 09:20:07] Perform umap nonlinear dimension reduction
#> ✔ [2026-07-02 09:20:13] Standard processing workflow completed
levels(pancreas_sub@meta.data[["SubCellType"]]) <- unique(
pancreas_sub@meta.data[["SubCellType"]]
)
pancreas_sub <- RenameClusters(
pancreas_sub,
group.by = "SubCellType",
nameslist = letters[1:8]
)
CellDimPlot(pancreas_sub, "newclusters")
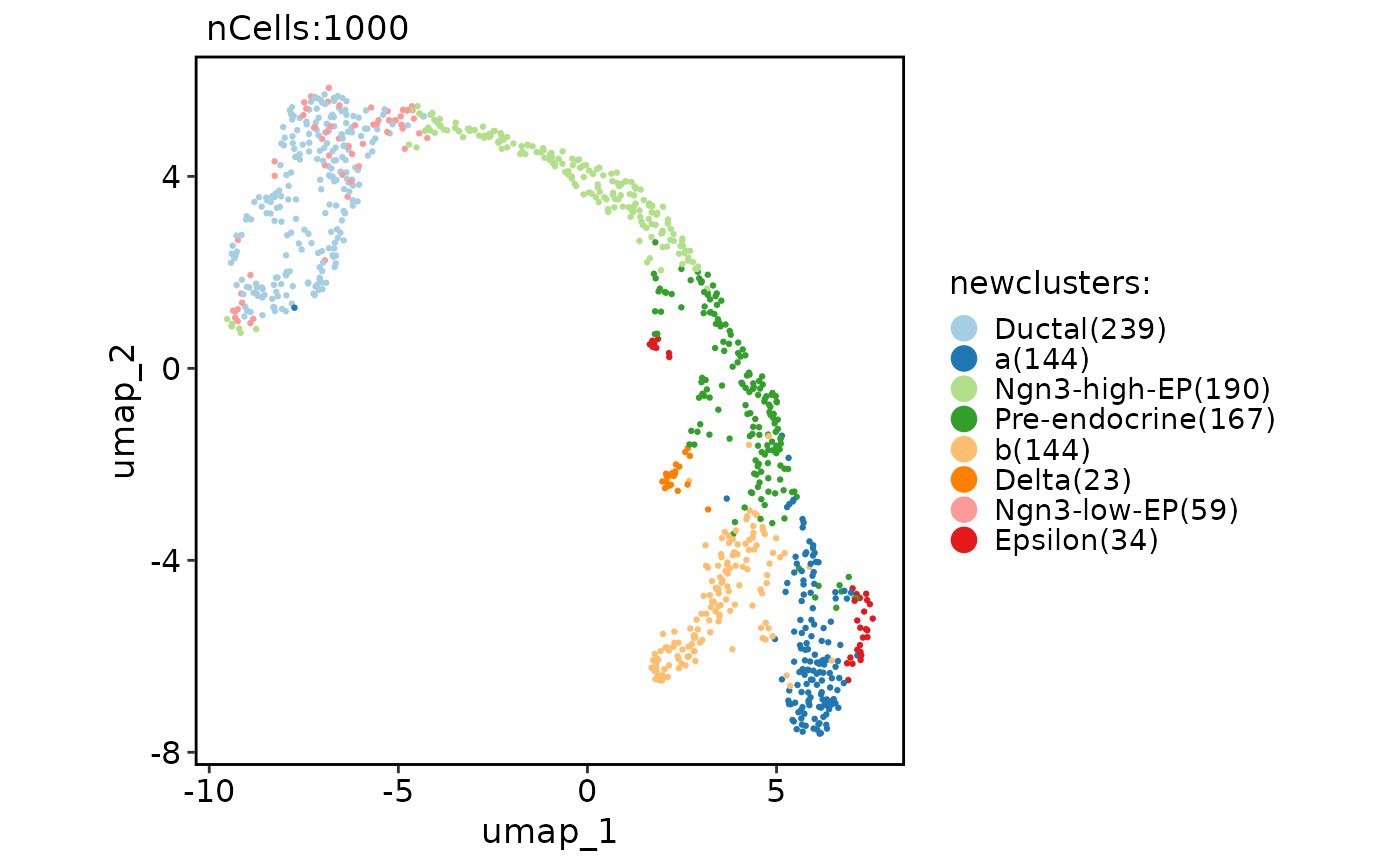
 # Rename specified clusters
pancreas_sub <- RenameClusters(pancreas_sub,
group.by = "SubCellType",
nameslist = list("a" = "Alpha", "b" = "Beta")
)
CellDimPlot(pancreas_sub, "newclusters")
# Rename specified clusters
pancreas_sub <- RenameClusters(pancreas_sub,
group.by = "SubCellType",
nameslist = list("a" = "Alpha", "b" = "Beta")
)
CellDimPlot(pancreas_sub, "newclusters")
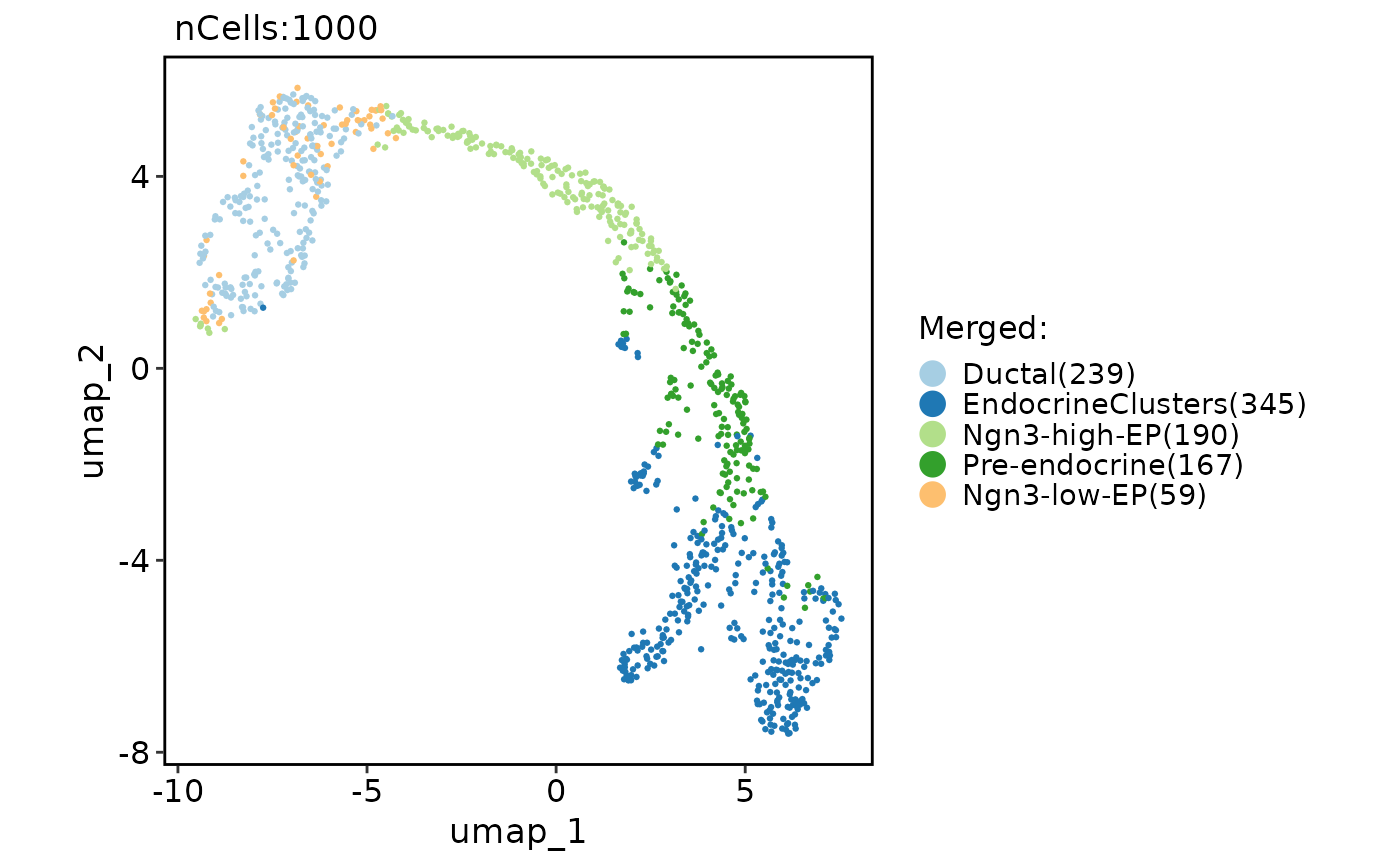
 # Merge and rename clusters
pancreas_sub <- RenameClusters(
pancreas_sub,
group.by = "SubCellType",
nameslist = list(
"EndocrineClusters" = c("Alpha", "Beta", "Epsilon", "Delta")
),
name = "Merged",
keep_levels = TRUE
)
CellDimPlot(pancreas_sub, "Merged")
# Merge and rename clusters
pancreas_sub <- RenameClusters(
pancreas_sub,
group.by = "SubCellType",
nameslist = list(
"EndocrineClusters" = c("Alpha", "Beta", "Epsilon", "Delta")
),
name = "Merged",
keep_levels = TRUE
)
CellDimPlot(pancreas_sub, "Merged")